Thromboembolism
Venous thromboembolism (VTE) is the joint term used mainly for two diseases: deep venous thrombosis (DVT) and pulmonary embolism (PE). Thrombotic disease was already known 2000 BC and the expression thrombosis was coined by Galenas around 150 AD. Thrombosis as a postoperative complication was not recognized until the end of the 19th century. During the 20th century a constant increase in frequency has been noted, due partly to an increase in the average life-span but also to sedentary habits in developed countries, and to factors which can influence the coagulability of the blood, the flow and the vessels. During the past 40 years, there has been considerable progress in the diagnosis, treatment, and prevention of venous thrombosis.
The three risk factors for VTE, referred to as Virchow’s triad, include reduced blood flow, damage to the vascular wall, and alteration of the blood components.
Venous thromboembolism is common in the US and kills more people than AIDS & breast cancer combined. VTE is the third most common vascular disease. Pulmonary Embolism occurs when the clot breaks loose and travels to the lungs, and is the most serious complication of VTE. PE is the leading cause of maternal death associated with childbirth. The incidence of PE is about 69 per 100,000 and results in approximately 200,000 deaths per year. The risk of recurrent DVT is high, and 30% suffer from post-thrombotic syndrome. Further, untreated PE carries a mortality rate of 30%.
VTE is a multi-factorial disease. Risk factors are divided into 3 categories: 1) physiological, 2) genetic, and 3) acquired. Physiological factors include age and hormonal influence like pregnancy or use of oral contraceptives. Acquired risk factors include malignancy, surgery, obesity, trauma, or prolonged immobility. Hereditary risk factors include deficiencies in anticoagulation proteins, increased levels of pro-coagulation proteins, and impaired function of proteins involved in hemostasis.
Deep venous thrombosis often does not present any clinical symptoms and is not discovered until secondary damage, such as circulatory disturbances and/or leg ulcers, appear. A mobile thrombus, however, can give rise to pulmonary embolism with dramatic clinical symptoms, often leading to death.
Thrombophilia
The term thrombophilia has been used to describe “the familial or acquired disorders of the hemostatic mechanism which are likely to predispose to thrombosis” (The British Committee for Standards in hematology). Prior to the discovery of blood coagulation inhibitors the importance of familial venous thrombophilia was not widely appreciated. During recent years, however, the term “thrombophilia” has been used more frequently instead of “thrombotic tendency”. With the discovery of anti-thrombin deficiency and later on during the eighties of protein C, protein S and factor XII-deficiencies the situation changed. Now approximately 25-30% of all cases of thrombosis could be explained by hereditary deficiency in coagulation inhibitors or hereditary defects in certain coagulation or fibrinolysis factors. But still the majority of thrombotic events were categorized as idiopathic venous thrombosis.
There occurred, however, a major change with the discovery of activated protein C resistance (APC resistance). At that time, up to 40% of additional cases with thrombophilia can be explained by this defect. The frequency of APC resistance in a general Caucasian population is estimated to be approximately 5 %.
Genetic risk factors in unselected thrombosis patients
| APC resistance | 20% |
| Prothrombin | 6% |
| Protein C | 3% |
| Protein S | 2% |
| Antithrombin | 1% |
| Plasminogen | 1% |
| Fibrinogen | 1% |
| Unexplained | 66% |
Diagnosis of thrombophilia
Thrombophilia usually presents as venous thrombosis or recurrent venous thrombosis in patients under the age of 45 years. These patients require a well-documented personal and family history and should also be considered for investigation. The decision to investigate thrombophilia should be made on an individual basis but patients with any of the following require special consideration.
Thrombophilic defects can be found in at least half the patients presenting with unprovoked VTE. Hereditary defects associated with an increased risk of VTE include deficiencies of anticoagulant proteins like Antithrombin, Protein C, and Protein S, as well as the Factor V Leiden mutation (which presents as a poor anticoagulant response to activated Protein C) and the Prothrombin G20210A mutation. Elevated FVIII is a dose-dependent risk factor, particularly for recurrent thrombosis. Acquired risk factors for thrombophilia include the presence of anti-phospholipid antibodies, malignancy, immobilization, and hormonal fluctuations.
The relative risk of a first occurrence of VTE in individuals with the above abnormalities varies from 2 – 11, with Antithrombin, Protein C, and Protein S deficiencies giving a relative risk of 7 – 10. Patients with Factor V Leiden mutation or prothrombin gene mutation have a slightly increased risk of recurrence after an initial episode of VTE, while those with mild hyperhomocysteinemia have a moderately increased risk, and those patients with consistently elevated FVIII levels, with lupus anticoagulant, or antiphospholipid antibodies have the highest risk of recurrence. Risk is further increased in patients with multiple defects. Idiopathic VTE, residual DVT on compression ultrasound, and persistently elevated D-dimer may also be indicative of risk of recurrence.
Due to the associated costs, routine screening of patients with VTE for underlying thrombophilic risk factors is impractical, and should be reserved for those patients where there is reason to believe may have an underlying predisposition for clot development, such as:
- Venous thrombosis under the age of 45 years
- Recurrent venous thrombosis or thrombophlebitis
- A family history of venous thrombosis
- A family history of a thrombophilic abnormality
- Venous thrombosis in an unusual site-e.g. mesenteric vein
- Spontaneous neonatal thrombosis
- Skin necrosis due to coumarin therapy
- Arterial thrombosis under the age of 30 years
- Thrombosis during pregnancy
Thrombophilia risk factors
Inherited Risk Factors
- APC resistance-Factor V Leiden
- Antithrombin deficiency
- Protein C deficiency
- Protein S deficiency
- Prothrombin G20210A Mutation
- Hyperhomocystenemia
- Elevated levels of FVIII, IX,XI
Acquired Risk Factors
- Age
- Malignancy, Hematologic Diseases
- Immobilization, Long-distance flights
- Trauma, Post-op
- Pregnancy, Estrogen Use
- Antiphospholipid Antibodies
Clinicians should be aware of thrombophilic disorders and be conscious of the need to investigate the patient groups described above. All patients with possible thrombophilia should have a full general medical assessment to exclude diseases which are associated with thrombosis such as malignancy, myeloproliferative disease, hyperlipidemia, and nephrotic syndrome.
If no such diseases are found then a thrombophilic disorder should be considered and the patient investigated for abnormalities.
Investigations should be extended to include key family members of patients found to have defects. If possible, parents, siblings and children of the propositus should be encouraged to attend for testing.
Anticoagulant therapy may make interpretation of these results difficult. Heparin therapy reduces the plasma levels of functional antithrombin and interferes with protein C assays. This seldom causes diagnostic problems since heparin is usually given for only short periods of time and the assay can be repeated after completion of heparin treatment. Coumarin therapy reduces the levels of protein C and protein S which are vitamin K dependent. Because coumarin is generally given on a long-term basis it frequently causes difficulties in the investigation of thrombophilia. It may be possible to make a tentative diagnosis of protein C or protein S deficiency in a patient on coumarin therapy if the ratio of protein C antigen or protein S antigen to factor II and/or factor X antigen is less than 1.0. To make a firm diagnosis it is necessary to test the patient after withdrawal of coumarin or during a period when heparin therapy has been substituted.
In summary, all patients with a possible thrombophilic abnormality should undergo the following studies.
- A clinical appraisal
- A family history appraisal
- A coagulation screen, including full blood count
- APC resistance
- Antithrombin
- Protein C
- Protein S
- Plasminogen
- Fibrinogen
- Fibrinolysis
- Antiphospholipid antibodies
Treatment of Thrombophilic Patients
An acute thrombotic episode should be treated conventionally with heparin followed by coumarin. Patients with antithrombin deficiency may be difficult to heparinize and require higher doses of heparin. Coumarin should be introduced slowly with no loading dose and overlapped with heparin for seven to ten days.
Patients should also be given general advice about minimizing the risk of thrombosis-dietary advice, cessation of smoking, avoiding periods of immobility. Women should, if possible, avoid synthetic estrogens and patients should be warned that they may require special treatment in situations of increased thrombotic risk such as trauma, surgery or pregnancy.
The management of patients with a thrombophilic abnormality and who have already suffered an episode of thrombosis, requires special consideration. Some authorities suggest that long-term anticoagulant prophylaxis is mandatory, even after a single thrombotic episode. Others suggest that this is unnecessary and consider that the risks of long-term anticoagulation outweigh the benefits. Patients not on long-term anticoagulants must be offered short-term prophylaxis to cover periods of increased thrombotic risk.
Since only a proportion of subjects with a heterozygous thrombophilic defect develop thrombosis, it is unjustifiable to put family members on antithrombotic prophylaxis solely on the basis of having a defect. It is, however, essential that these asymptomatic members are carefully counselled with respect to their defect and offered short-term prophylaxis in special situations of extra thrombotic risk.
Patients and their families must be aware of the possible requirement for additional therapy in situations where the risk of thrombosis is increased, e.g. trauma, surgery and pregnancy.
Pregnancy presents special problems. Women who have already had a thrombosis and who have a known defect require anticoagulation throughout pregnancy-usually using dose-adjusted subcutaneous heparin. Women who have had no previous thrombosis but are known to have a defect require individual consideration. Those who are anti-thrombin deficient require anticoagulation from conception onwards, but those who have other heterozygous defects may, in some cases, not require anticoagulation until later in pregnancy.
All women with thrombophilic conditions should be offered thromboprophylaxis in connection with delivery and in the puerperium.
In recent studies, low doses of low molecular weight heparin and/or low doses of coumarin derivatives have been investigated in patients who need prophylactic or long-term treatment. Hitherto the results are encouraging.
APC Resistance
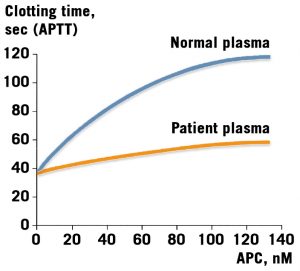
APC resistance, an autosomal dominant hereditary defect, represents the most common hereditary blood coagulation disorder identified so far.
The original observation was made on a male patient, born in 1942 and with multiple episodes of deep venous thrombosis. The patient’s poor anti-coagulant response to added APC is shown.
Recently, data have been published which suggest that also unactivated factor V serves as an APC cofactor.
APC resistance is present in 5% of a nor-mal population and approximately 40% in patients with thrombosis; by the age of 50 about 40% of these have experienced a thrombotic episode.
Since the discovery of APC resistance in 1993 by Dahlback et al, the findings have been confirmed by numerous research centers in both Europe and the USA.
It is very tempting to believe that this mutation in the factor V gene has had an evolutionary advantage. Previously when bleeding was the only hemostatic threat to life, it was probably an advantage to be somewhat hypercoagulable. Nowadays, with a longer average life-span, it has, instead, turned into a risk factor for thrombosis.
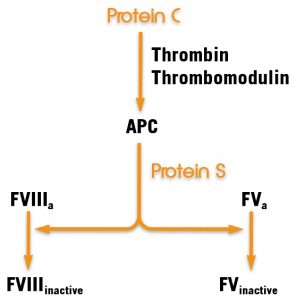
The main cause of APC resistance is a point mutation in the coagulation factor V gene. This mutation results in an amino acid change from arginine to glutamine. It occurs at a site which corresponds to position 506 in the activated factor V (=FVa), a cleavage site for APC, and prevents normal degradation of factor Va by APC.
The known role of protein S as a cofactor to APC is in this reaction as well as in the degradation of FVIIIa. Down regulation of coagulation through inactivation of FVIIIa and FVa by the protein C pathway.
Future Aspects
Many questions remain to be answered: does the high frequency of APC resistance justify treatment with anticoagulants in thrombophilic persons? What is the cost-benefit situation with respect to routine investigations of families with APC resistance?
Coagulation Cascade
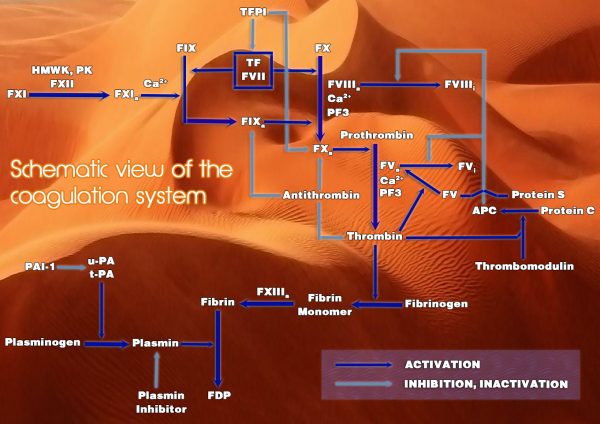
Assays for Diagnosing Thrombophilic Patients
There are currently no global assays available to diagnose thrombophilia, so a panel of assays is performed. Suggested testing varies between experts.
Thrombophilia Screening Panel
Diapharma has assay kits and reagents for thrombotic risk assessment.
- Chromogenix Coatest® APC Resistance V and VS: Validated, FDA cleared kit comes in two sizes to suit your needs. The unsurpassed sensitivity for the FV:Q506 mutation provides genotype information without the hassles of genetic testing, and is more convenient and cost-effective with faster turn-around time than PCR
- Chromogenix Coamatic® Antithrombin: Use of FXa instead of thrombin eliminates interference from heparin cofactor II and thrombin inhibitors. The assay also allows for accurate determination in patients receiving heparin therapy. Applications are available for a wide range of automated coagulation analyzers, and the highly precise kit gives CV values of less than 5%.
- Chromogenix Coamatic® Protein C: The chromogenic assay is the method of choice for detecting Protein C deficiency. Coamatic® Protein C provides simple, rapid, and reliable results, and is adapted to a broad range of instruments.
- Chromogenix Coamatic® Factor VIII: An ideal in vitro diagnostic tool for measuring pre- and post-treatment patient FVIII activity levels. The assay is economical and fast, with protocols available for most automated coagulation analyzers.
Laboratory Evaluation of Thrombotic Risk
| Risk Factor | Laboratory Assay |
| Antithrombin Deficiency | Antithrombin activity |
| Protein C Deficiency | Protein C activity (chromogenic) |
| Protein S Deficiency | Free Protein S Antigen (ELISA, LIA) |
| APC Resistance / Factor V Leiden Mutation | APC Resistance (clotting); FV Leiden genetic test if abnormal |
| Prothrombin Mutation G20210A | Genetic Test |
| Hypherhomocysteinemia | EIA, HPLC |
| Elevated Factor VIII Activity | Factor VIII activity (clotting or chromogenic) |
| Lupus Anticoagulant | DRVVT Clotting Assay |
| Anticardiolipin Antibodies | aCL IgG/IgM Antigen ELISA |
Treatment and Monitoring of Thrombophilic Patients
Patients with idiopathic VTE have a risk of recurrence of approximately 7 – 10% per year once anticoagulant therapy is stopped. Longer duration of anticoagulant therapy may be beneficial; however, limitations on currently available anticoagulants can make long-term therapy problematic.
An acute thrombotic episode is typically treated with heparin, either unfractionated heparin (UFH) or low molecular weight heparin (LMWH), followed by longer term warfarin therapy.
Heparin is a naturally occurring polysaccharide used for the treatment and prevention of thrombotic diseases. Heparin works with Antithrombin to FXa and FIIa (thrombin). Because LMWH inhibits FXa to a greater extent than FIIa, an anti-Xa chromogenic assay is recommended when monitoring is necessary.
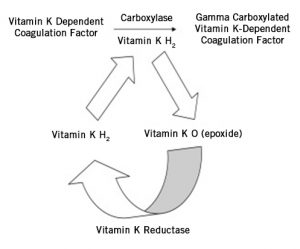
Warfarin is an oral anticoagulant that acts as a vitamin K antagonist, suppressing the production of the vitamin K dependent coagulation factors such as FII, FVII, FIX, and FX. Full anticoagulation potential occurs approximately 5 – 10 days after warfarin therapy begins. Warfarin has a narrow therapeutic window, and the risk of major bleeding with coumarin derivatives like warfarin is 2% per year. Warfarin is typically monitored with the PT/INR, or with a chromogenic Factor X assay when inhibitors like lupus anticoagulant are present.
Because of the limitations of current anticoagulants, new drugs are being developed for the prevention and treatment of VTE.
FXa Inhibitors
FXa is an attractive target for new anticoagulants, and much research has been focused on compounds that act as inhibitors to FXa as potential new antithrombotic drugs. Indirect FXa inhibitors, like fondaparinux and idraparinux, are mediated through their binding to and activation of antithrombin, which inhibits FXa. Fondaparinux can be given once daily, is likely to be a safe and effective anticoagulant in patients with HIT, and, like LMWH and most of the FXa inhibitors, does not normally need laboratory monitoring. Direct FXa inhibitors, in contrast, inhibit FXa without the need for antithrombin. A number of direct FXa inhibitors, like Razaxaban, are currently being evaluated and undergoing clinical trials.
Direct Thrombin Inhibitors
Direct thrombin inhibitors (DTIs) inhibit thrombin by directly binding and inactivating thrombin. DTIs like Argatroban produce a more predictable anticoagulant response than heparin, and do not cause HIT. DTIs like Ximelagatran also may offer some benefits over warfarin. They do not act through inhibition of vitamin K, and are therefore not affected by diet like warfarin, have lower potential for drug interactions, and have a larger therapeutic window, so that routine laboratory monitoring and dose adjustment is unnecessary.
There are also disadvantages to these new anticoagulants. Antidotes are not always available, they are more expensive than traditional anticoagulants, and the safety and efficacy is not well-established for some. The ultimate utility of these new anticoagulants is promising, however, and requires further clinical trials.
Anticoagulant Monitoring
The following assay kits and reagents are available from Diapharma for anticoagulant monitoring:
- DiaPharma Factor X
Chromogenic assay for monitoring patients on oral anticoagulant therapy with unreliable INRs, due to interference from lupus inhibitors, and when bridging Argatroban® patients to warfarin.
- Chromogenix Coamatic® Heparin
Chromogenic assay for measuring anti-Xa activity of heparin.