Von Willebrand disease (VWD) is the most common inherited bleeding disease and is caused by primary hemostasis defects. VWD is classically characterized by frequent nosebleeds, easy bruising, heavy and long-lasting menstrual periods, prolonged bleeding caused by wounds or medical procedures, and bleeding in mucous membranes such as the gums and gastrointestinal linings. In severe circumstances, musculoskeletal bleeding can occur.
VWD is caused by defective or deficient von Willebrand factor (vWF) protein. vWF is an important component of primary hemostasis by facilitating platelet adhesion to damaged vascular endothelium. vWF mediates clotting by binding exposed subendothelial collagen through domains A1 and A3 (Figure 1). Once bound to the subendothelial connective tissue, vWF mediates platelet adhesion under rapid blood flow conditions. High shear stress is required for the A1 domain of vWF to bind the GPIbα glycoprotein domain of platelets, thereby tethering platelets to the subendothelium and mediating platelet adhesion (Figures 1 and 2A). Platelet adhesion is stabilized through interaction with collagen receptors. This facilitates platelet activation, aggregation, and platelet plug formation, also called a platelet thrombus. vWF contributes to fibrin clot formation through interaction with coagulation factor VIII (FVIII) (Figure 1). vWF protects FVIII from degradation and chaperones FVIII to the site of exposed subendothelium. Stabilized FVIII then proceeds to function as a key co-factor during secondary hemostasis.
An interesting aspect of vWF is that vWF function is dependent upon the size of vWF multimers. vWF multimers substantial enough to bind platelets undergo structural conformations that expose the ADAMTS-13 cleavage site in domain A2. The ADAMTS-13 metalloprotease specifically cleaves vWF multimers to prevent the accumulation of ultra-large multimers. ADAMTS-13 cleavage alters the distribution of vWF within the blood, transforming large multimers into smaller multimers in a well-characterized pattern. The multimer distribution within the blood indicates the balance of multimer assembly, removal from circulation, and cleavage by ADAMTS-13.
Overall, the three functional activities of vWF are to bind platelets, bind collagen, and bind FVIII. Variations to vWF, including multimer dynamics, can interfere with platelet binding, collagen binding, and FVIII stability, ultimately leading to VWD.

Figure 1: von Willebrand factor (vWF) contains platelet GPIbα-binding sites at domain A1, collagen-binding sites at domains A1 and A3, and Factor VIII (FVIII) domains at D’-D3.
Diagnosis
The diagnosis of VWD is challenging due to variability in vWF levels, vWF activity, and symptoms associated with it. VWD is carefully organized into 3 over-arching types: Type 1, Type 2, and Type 3. Type 1 and Type 3 are similar in that they are classified as quantitative deficiencies in vWF, meaning that vWF levels are low or absent from the blood.
VWD Type 1 is the most common and mild variety of VWD. Type 1 is characterized by low levels of vWF, decreased vWF-platelet binding activity, reduced vWF-collagen binding, and reduced levels of FVIII (Figure 2B). Symptoms may include mucocutaneous bleeding, occasional increases in bleeding following trauma or menstruation, and bruising.
VWD Type 3 is the most severe form of VWD. vWF and factor VIII levels are very low or undetectable in the blood (Figure 2C). Symptoms associated with VWD Type 3 can include severe mucocutaneous bleeding, musculoskeletal bleeding, and the development of hematomas.
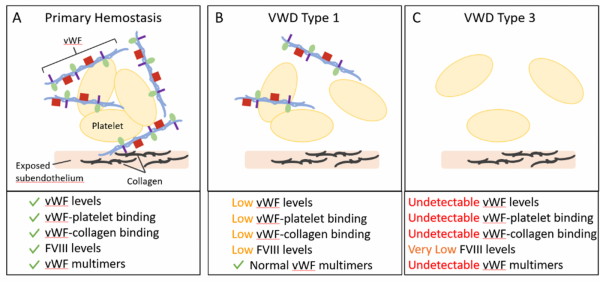
Figure 2: (A) Primary Hemostasis requires multiple components, including von Willebrand factor (vWF), platelets, Factor VIII (FVIII), and collagen. (B) Von Willebrand Disease (VWD) Type 1 is a partial quantitative deficiency in vWF with decreases in vWF levels. (C) VWD Type 3 is a complete quantitative deficiency in vWF with undetectable levels of vWF. VWD Type 1 and Type 3 display deficiencies in primary hemostasis that can lead to bleeding.
VWD Type 2 accounts for approximately 20% of VWD cases. VWD Type 2 typically has normal levels of vWF protein in the blood and instead displays defects in vWF activity. Type 2 is classified as a qualitative deficiency in vWF functionality, unique from the reduced levels of vWF described in VWD Types 1 and 3. VWD Type 2 is further separated into phenotypic subtypes: Type 2A, 2B, 2M, and 2N.
Type 2A is characterized by vWF proteins that are unable to bind platelets, and therefore platelets are unable to aggregate to form a clot. Mutations associated with Type 2A affect the ADAMTS-13 cleavage site, such that proteolysis is increased (Figure 3A). Increased ADAMTS-13 cleavage inhibits the formation of vWF multimers. Defective multimer assembly may also drive abnormal vWF multimers. Type 2A is associated with mild to moderate mucocutaneous bleeding.
Type 2B is unique in that it is characterized by a gain of vWF function. In Type 2B, vWF binds platelets with increased affinity due to mutations in the binding site for platelet GP1b. This binding of mutant vWF to platelets inhibits the activation and adhesion of platelets in response to endothelial injury (Figure 3B). Elevated destruction of vWF-platelet complexes may cause decreased levels of platelets in the blood, a condition known as thrombocytopenia. In addition to increased risks for thrombocytopenia, individuals affected with Type 2B may experience mild to moderate mucocutaneous bleeding.
Type 2M is similar to Type 2A in that vWF is unable to bind platelets. However, the loss of function seen in Type 2M is due to the inability of vWF to bind platelet receptor GP1bα (Figure 3C). This dysfunction is suggested to be due to misfolding of the vWF protein. These defects lead to decreased platelet adhesion and defects in collagen-binding. Individuals affected with Type 2M may experience mild to moderate mucocutaneous bleeding, and severe bleeding in some cases.
Type 2N is characterized by an inability of vWF to transport FVIII to the site of endothelial injury due to mutations in the D3 domain containing the FVIII binding site (Figure 3D). Without the stability of vWF to form a vWF-FVIII complex, FVIII is degraded resulting in reduced levels in the blood. Individuals affected with Type 2N may experience severe bleeding following trauma or surgery. Due to its association with reduced FVIII functionality, Type 2N can resemble hemophilia A, which is a bleeding disorder characterized by deficient FVIII.
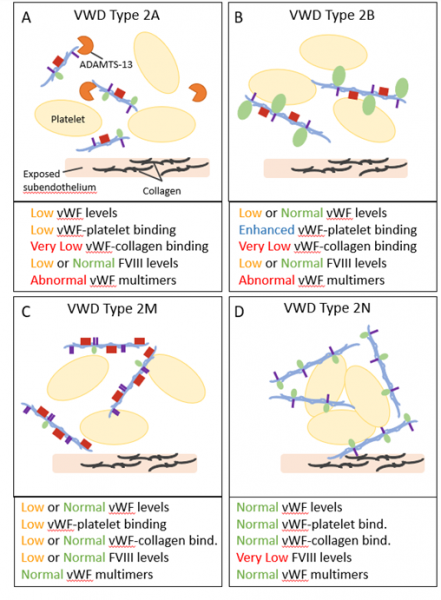
Figure 3: Von Willebrand Disease (VWD) Type 2 is categorized as qualitative deficiencies in vWF functionality. Type 2 is further categorized into Type 2A, 2B, 2M, and 2N subtypes. (A) Type 2A is characterized by vWF that is unable to bind platelets due to increased ADAMTS-13 proteolysis and abnormal vWF multimer formation. (B) Type 2B contains mutant vWFs that bind platelets with increased affinity. (C) Type 2M is characterized by vWF proteins that are unable to bind platelets through the platelet receptor GP1bα. (D) Type 2N is characterized by an inability of vWF to bind and chaperone Factor VIII (FVIII).