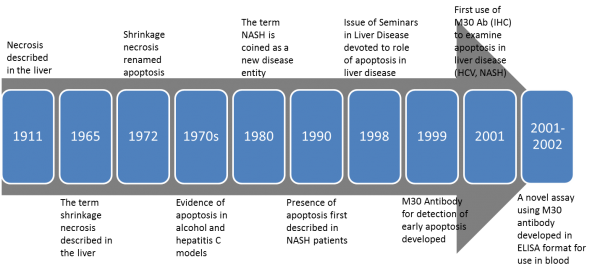
An association between liver disease, keratins, and liver cell death has been known for over a hundred years. In fact, in 1911 F.B. Mallory observed that “The so-called alcoholic type of cirrhosis is characterized by a peculiar hyaline degeneration of the cytoplasm of the liver cells preceding necrosis.” Alcoholic hyaline bodies that occur characteristically in abundance in the diseased livers of alcoholic subjects became known as Mallory bodies (MBs) and Mallory-Denk bodies (MDBs), which were eventually found to be characteristics of other liver diseases as well.
In the 1960s, a different type of cell death was first described in hepatocytes as shrinkage necrosis (“…might have been due to gradual autodigestion of cytoplasm…”). This term was first described in the rat liver in 1965 following induction of liver damage. It wasn’t until 1972 before the term apoptosis was proposed by John Kerr and his colleagues, who later proposed that the type of hepatocyte cell death observed in subjects with chronic hepatitis C was apoptosis. Also in the 1970s, Walker et. al described the cytotoxic effects of alcohol on liver cells, and eventually evidence of apoptosis was shown in a variety of models due to the effects of alcohol on liver cells.
It was known that K8 and K18 are among the essential components of MDBs. The critical important of these structural proteins was confirmed in animal and human studies. Keratin variants and associated disease is now well known. Approximately 4% of the white individuals carry K8/K18 mutations that predispose them to development and adverse outcomes in liver diseases. Animal models support the importance of K18 in human disease show their cytoprotective role in preventing liver injury.
Apoptosis is known to play a critical role in both physiological cell death and its dysregulation in many types of disease. As described above, a variety of models highlighted a role in liver disease, as in ASH and Hepatitis C. As the cell death field evolved (and continues to evolve) in terminology, so did the hepatology field. The term NASH was first proposed as a new disease entity in 1980 but the close similarities between NASH and ASH had been immediately recognized. Although the mechanisms may not have been known, evidence of apoptosis was first observed in 1990 in liver biopsy specimens from NASH subjects.
As tools for studying cell death continued to emerge with mechanistic discoveries in the field, applications of those tools by scientists practicing in the art in the liver field emerged as well. In 1998, an entire issue of Seminars in Liver Disease was devoted to the role of apoptosis in liver disease. In 1999, an antibody to detect early phases of apoptosis as compared to other methods (ie, TUNEL or annexin V) was developed. Naturally, this new apoptosis technique was applied to the liver in 2001 and apoptosis was observed in HCV as well as in NASH biopsies. Also in 2001 Denk et.al, who recognized apoptosis as occurring in both ASH and NASH, observed that disturbance of the keratin system (keratin 8 and keratin 18) may significantly contribute to cell damage. Keratin antibodies have been used for many years but with the discovery of a novel antibody that recognized a product only generated during caspase activation in apoptosis, a new research tool emerged.
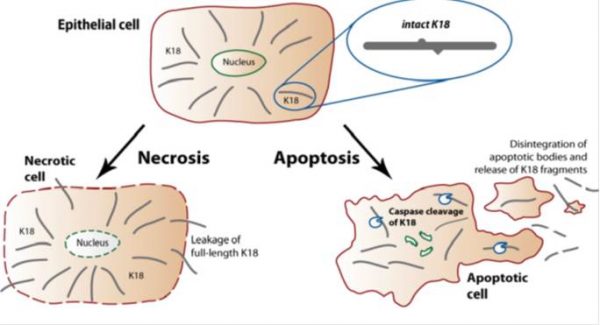
During apoptosis, keratin filaments are altered and remarkably stable keratin fragments are generated. Activation of caspases leads to proteolysis of numerous cellular proteins, including structural components of epithelial cells. K18 (also referred to in the literature as CK18) along with Keratin 8 are the major components of intermediate filaments in simple epithelial cells, and the only keratin pair in normal hepatocytes. K18 is often used to identify differentiated isolated hepatocytes. During apoptosis, K8/K18 fragments are dramatically reorganized and K18 is cleaved by caspases at multiple sequence sites. Cleavage of K18 is an early event occurring during apoptosis. The M30 antibody recognizes a neo-epitope exposed after caspase cleavage of K18 after the aspartic acid residue 396. Cleavage at this position occurs early during apoptosis by caspase 9 and during the execution phase by caspase 3 and caspase 7.