Protein S
Protein S (S-Protein) is a vitamin K-dependent plasma glycoprotein synthesized in the endothelium. Protein S exists in two forms: a free form and a complex form bound to complement protein C4b-binding protein (C4BP).
Free Protein S plays a role in the anti coagulation pathway, where it functions as a cofactor to Protein C in the inactivation of Factors Va and VIIIa.
Protein S deficiency is a rare blood disorder which can lead to an increased risk of thrombosis.
Protein S, an important regulatory protein in hemostasis
Protein S is a vitamin K-dependent protein which has an important role in regulation of blood coagulation. This has been deduced from the serious clinical conditions which occur in individuals with severe protein S deficiency.
At the same time it should be recognized that the exact mechanism of action of protein S in vivo has yet to be elucidated. It is well established, that only free protein S has anticoagulant activity however and not its complex with C4b-binding protein. Herein we review the biochemistry of protein S and clinical findings related to protein S deficiency.
Hemostasis is a fine-tuned balance between procoagulant events and their regulation by anticoagulant systems and by fibrinolysis. At any moment there is evidence of on-going activation of all three systems, albeit on a very low level. Hemostasis is thus a dynamic process.It is generally agreed that coagulation is triggered in vivo by the exposure of tissue factor to blood and the ensuing complexing with and activation of factor VII. The tissue factor – factor VIIa complex then activates factor IX and factor X whereafter thrombin generation and fibrinogen cleavage occurs. Coagulation factors VIII and V serve as very important cofactors in the activation of factor X and prothrombin, respectively. Each cofactor provides a 1000-fold stimulation.
The Coagulation Cascade
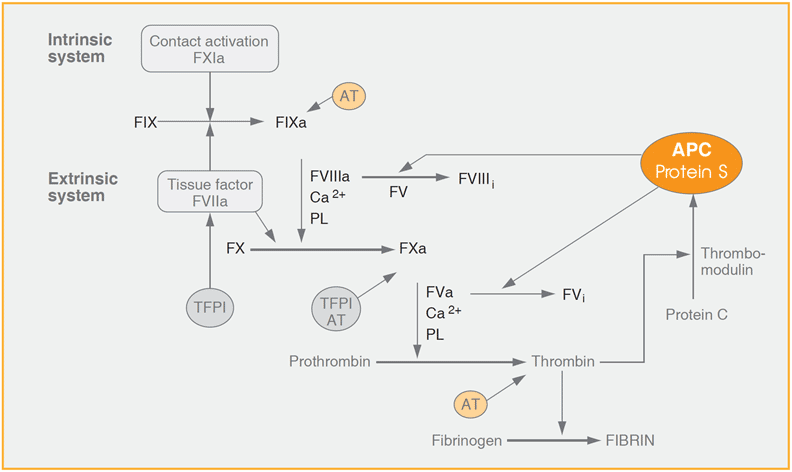
Blood coagulation can somewhat simplified be described as a cascade system of proteolytic reactions initiated in response to tissue damage. In each reaction, an inactive zymogen is converted to its active enzyme counterpart, which then participates in the subsequent step of the coagulation cascade. The protein C anticoagulant pathway is the key system for down regulation of blood coagulation. Here, protein S serves as a cofactor to APC in the inactivation of coagulation factors VIIIa and Va. In addition to its cofactor activity to APC, protein S also has an anticoagulant activity that is independent of APC.
Coagulation is down-regulated by the protein C anticoagulant pathway, in which activated protein C (APC) degrades activated factors VIII and V through specific proteolytic cleavage. Protein S is a cofactor to APC in these events. Protein S also seems to have anticoagulant activities which are independent of APC.
Already formed fibrin is later degraded by plasmin, the key enzyme in the fibrinolytic system. The importance of the protein C anticoagulant pathway has been amply demonstrated through the serious thrombotic events which occur in connection with severe deficiency of protein C and protein S. It has also been demonstrated by the 7-fold increase in thrombotic risk which is associated with one commonly ocurring single point mutation in the factor V gene. This mutation results in impaired degradation of factor Va at one of the three APC cleavage sites, a condition denoted as APC resistance. One intriguing fact regarding protein S is that it circulates in plasma partly (about 60%) in complex with C4b-binding protein (C4BP); extensive research has clearly shown that only free protein S has anticoagulant activity.
It must be emphasized that it is not yet known which anticoagulant activity of protein S is most important in vivo, and its detailed mechanism of action awaits clarification.
Due to difficulties in determination of protein S antigen and activity, there is so far no firm knowledge of protein S deficiency among the general population; and hence the increased risk connected with protein S deficiency is not easily assessed. The available data indicate, along with other anticoagulant components, that a clinically important risk connected with fairly mild (heterozygous) protein S deficiency is primarily found in connection with other inherited or acquired thrombotic risk factors.
Protein S Biochemistry
Basic structural and functional aspects
Protein S is a vitamin K dependent plasma glycoprotein with a molecular weight of approximately 70 kDa and it circulates in plasma at a concentration of 25 μg/mL with a half-life of roughly two days. About 40% of Protein S in plasma is in a free form whereas 60% is in complex with C4b-binding protein (C4BP). Free Protein S functions as a cofactor to APC in the APC-dependent degradation of factor Va and factor VIIIa.
It is synthesized not only in the liver but also at other sites such as in endothelial cells, in Leydig cells in the testis and in megakaryocytes, the latter resulting in presence of protein S in platelets at low concentrations. Protein S is synthesized as a precursor protein but a leader sequence is cleaved before secretion of the mature protein, containing 635 amino acids.
Gene complexity
A finding which has complicated the search for mutations is that there are two highly homologous protein S genes in the human genome, both located on chromosome 13. Only one of them is active and thus expresses protein S (PSa) whereas the second gene is a pseudogene (PSb), which lacks the first of 15 exons of PSa.
Domain structure
Protein S is a multimodular protein, containing, in common with other vitamin K dependent proteins, a g-carboxy-glutamic acid domain (Gla-domain ) and an epidermal growth factor like domain (EGFdomain). It also contains a thrombin sensitive region and a carboxyterminal region which is strongly homologous to the sex hormone binding globulin (SHBG). The SHBG-like domain substitutes for the serine protease domain present in factors VII, IX, X, prothrombin and protein C. Thus, protein S is not a serine protease but a regulatory protein.
Schematic representation of human protein S
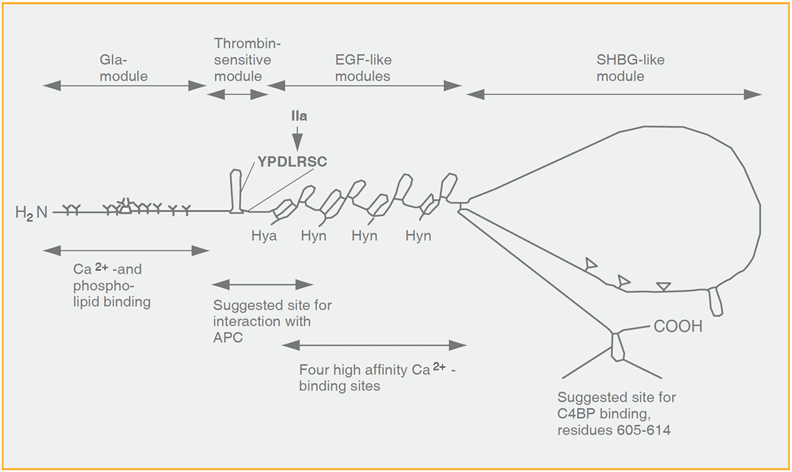
Protein S is composed of different modules; the Gla-domain, the thrombin-sensitive region, four EGF-modules and a region which is homologous to the sex hormone binding globulin (SHBG-like domain) (Reproduced by permission of Prof. Björn Dahlbäck)
The EGF domain contains four EGF-like modules, each containing either one b-hydroxy-Asp (Hya) or one b-hydroxy-Asn (Hyn). Calcium ions bind to the Hyn-containing EGF-like modules with a very high affinity. The Gla-domain is crucial in anchoring protein S to phospholipid membranes.
The affinity for phospholipids is highly increased after binding of multiple calcium ions; a conformational change occurs which is mandatory for the APC cofactor activity of protein S.
The thrombin-sensitive region contains one disulphide bond and is located between the Gla- and the EGFdomains. Protein S is considerably more sensitive to cleavage in the absence of calcium ions, a fact of importance for maintaining the native state during purification. After cleavage by thrombin, the Gladomain remains linked to the rest of the protein S molecule via the disulphide bond.
However, the APC cofactor activity is lost since the thrombin-cleaved protein no longer features the necessary calcium-dependent conformational change. Cleavage in the thrombin-sensitive region and loss of cofactor activity has also been reported to be effected by factor Xa.
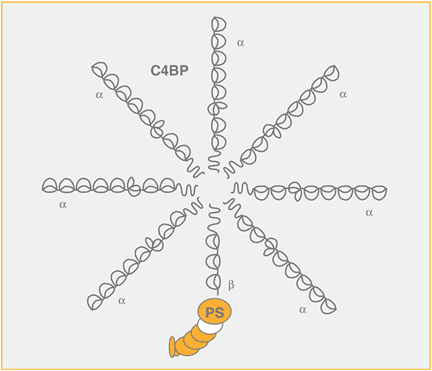
Interaction with C4BP
C4BP is an octopus-like molecule which contains seven identical a-chains (MW 70 kDa) and a smaller b-chain (45 kDa). There is one binding site for C4b in each of the a-chains, whereas Protein S binds to the b-chain.
The affinity of protein S to C4BP is several hundredfold increased by calcium ions (Kd approximately 5 x 10-10 mol/L) and this high affinity prevails in plasma where the free calcium ion concentration is 2 mmol/L. The level of free protein S corresponds to the molar excess of b-chain containing C4BP. Similar to C-reactive protein and fibrinogen, C4BP is an acute phase reactant, and its concentration in plasma may increase as much as four-fold. Does this result in free protein S levels approaching zero? Actually not, since C4BP exists in two forms: Under normal circumstances, about 10-15% of the C4BP circulating in plasma lacks the b-chain and hence does not bind protein S. During an acute phase reaction, primarily the species of C4BP which lacks the b-chain is increased and therefore no significant decrease of free protein S occurs.
Interaction with APC
Only the free, native form of protein S binds to APC and functions as a cofactor. Protein S has the highest affinity for negatively charged phospholipids of all the vitamin K-dependent proteins and, has been shown to increase, by approximately 10-fold, the affinity of APC for membranes or vesicles containing such phospholipids.
Degradation of FVa and FVIIIa by activated protein C
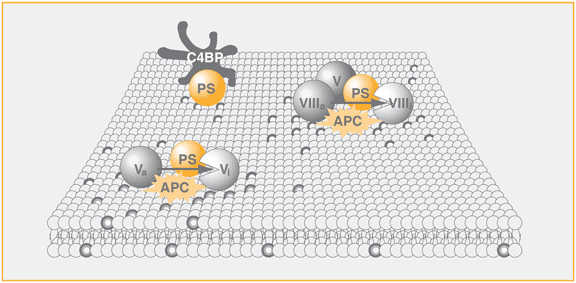
Protein S acts as a cofactor to activated protein C (APC) in the degradation of FVa and FVIIIa. The cofactor activity of protein S to APC is expressed by increasing the affinity of APC for phospholipids and relocating the active site of APC closer to the membrane surface that contains the activated coagulation factors. In the inactivation of FVIIIa, FV and protein S act as synergistic cofactors to APC.
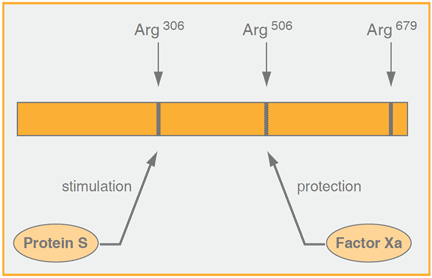
This may be of physiological importance since APC degrades preferentially membrane-bound factor Va and factor VIIIa but not the circulating, unactivated cofactors. No circulating complex in plasma of Protein S and APC has been detected, which may be taken as support that the cofactor role of protein S is linked to membranes. Apart from increasing the affinity of APC to membranes, protein S also enhances the rate of cleavage of Arg306 in factor Va by APC and it works in concert with factor V to increase the ability of APC to inactivate factor VIIIa. It has been shown that upon binding of protein S to APC, the distance from the active site of APC to the membrane surface is decreased about 1 nm and this decrease may be reponsible for the increased cleavage rate of Arg 306 in factor Va. Furthermore, protein S abrogates the protective effects against APC-dependent inactivation of factor Va by factor Xa and similarly of factor VIIIa by factor IXa.
APC-independent anticoagulant activity of protein S
Protein S has been shown to exert a direct inhibitory effect on the prothrombinase complex, later shown to be due to binding to factor Xa and to factor Va in purified systems, thereby impairing prothrombin activation. Results supporting a direct anticoagulant role for protein S were obtained under flow conditions in the presence of endothelial cells, where increased thrombin generation was noticed with protein S deficient plasma.
Not surprisingly, an interaction with a negatively charged phospholipid surface seems to be a prerequisite for the anticoagulant effect.
It is clear from the above that in vitro studies indicate several roles for protein S in regulation of hemostasis. It should be emphasized, though, that it is not yet known which is the most important mechanism of action in vivo for protein S either as a cofactor for APC or as a direct anticoagulant.
For a more detailed discussion on the structure and function of protein S related to hemostasis, the reader is referred to a comprehensive review on the protein C anticoagulant pathway
Clinical Aspects of Protein S
Involvement of protein S in areas other than hemostasis
In its complex with protein S, C4BP still has normal binding capacity for C4b to its seven a-chains and hence retains its biological activity. Protein S may serve as a regulator during local inflammatory events, since it has been shown that the C4BP-protein S complex binds to negatively charged phospholipid membranes, the binding being mediated by protein S.51 Thus, the directing of C4BP via protein S to perturbed cells with exposed negatively charged phospholipids may promote local down-regulation of inflammatory reactions.
Another interesting aspect of a tentative protein S function is in the area of cell proliferation. It has been shown that protein S binds to the tyrosine kinase receptor denoted Rse/Tyro 3 and to a receptor on smooth muscle cells. Upon binding, protein S induces phosphorylation and stimulates a mitogenic response.
This effect is not only confined to protein S among the coagulation factors, since it is known that this is one of the many roles of thrombin. Later, also factor Xa and protein C/APC have been shown or suggested to be involved in cellular activation through their binding, respectively, to effector protease receptor 1 (EPR1) and to an endothelial protein C receptor (EPCR). Indeed, it may therefore be possible that the above described direct binding of protein S to factor Xa may be more relevant in cell proliferation and inflammation than in hemostasis. Also, the expression of protein S in many different cell types may also be related to such areas.
In summary, research findings during the 1990’s have not only unravelled much beauty within the protein C anticoagulant pathway but also indicated a complex interplay between this system and inflammation and cell proliferation.
Role for protein S in areas other than hemostasis
- Inflammation: PrS / C4BP binds to negatively charged phospholipids, such as on perturbed cells
- Cell proliferation: Protein S binds to a tyrosine kinase receptor (Rse/Tyro 3) and to a receptor on smooth muscle cells-induces mitogenic response
Variation of protein S levels in plasma
The level of protein S in plasma is influenced in several ways. Being a vitamin K-dependent protein, its concentration is decreased during treatment with oral anticoagulants. With a half life of two days, similar to prothrombin, the rate of decrease for protein S is much lower than for protein C and factor VII which have half-lives of about 7 hours.
Realizing the large differences between various methods for determining total and free protein S antigen and (free) protein S activity, and also recognizing their inherent complexity, somewhat different normal ranges have been reported. It is reasonable to state, that a representative normal range is 70 – 140% for total protein S. Considering 25 μg/mL as the mean concentration, this corresponds to a range of 15 – 35 μg/mL. A number of studies have shown that the level of protein S is influenced by sex hormones, most probably estrogens. Thus, premenopausal women have lower values than men and postmenopausal women. Significantly lower mean values of total and free protein S are obtained in pregnant women (from 25 μg/mL to 15 μg/mL) and in women using oral contraceptives (from 25 μg/mL to 18 μg/mL). It is well known that women enter into a hypercoagulable state during pregnancy, but thrombotic events are rare. It may, however, not be excluded that women who develop phospholipid antibodies have a greater thrombotic risk if they have low protein S levels before pregnancy.
Not surprisingly, as for most hemostatic factors, protein S levels are also low in neonates. Due to quite low levels also for C4BP, the decrease of free, active protein S is less pronounced, however, and the impact on the hemostatic balance may be limited.
Influences on Protein S levels
- Increase with age
- Decreases during pregnancy and use of oral contraceptives
- Decreases during DIC, liver disease and in chronic inflammatory bowel disease
- Decreases during anti-vitamin K therapy.
Protein S deficiency
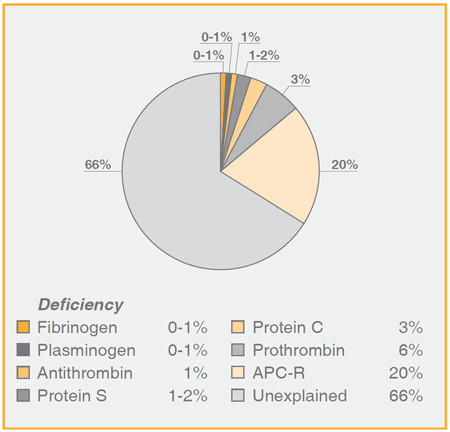
Although the exact role of protein S in vivo in the protein C anticoagulant pathway yet has to be clarified, there is no doubt that protein S is an important anticoagulant protein and that protein S deficiency is primarily associated with venous thromboembolism. The first thrombotic cases with protein S deficiency linked to familial thrombophilia were reported 1984 and rapidly confirmed. Severe protein S deficiency resulting from the extremely rare homozygosity for this trait is associated with serious thrombotic complications and purpura fulminans early in life. The reported prevalence of protein S deficiency in thrombosis subjects varies between 1.5 to 7%, the difference being due to methods used and to the selection of subjects. However, it is not possible to assess a relative risk increase for thromboembolism with protein S deficiency, since its prevalence in the general population is not known. Investigations on large population cohorts have so far been hampered due to the lack of reliable methods which also are suitable for screening purposes. From a large study involving close to 500 consecutive subjects and 500 controls it seems that isolated protein S deficiency may be only a mild risk factor.
Clinical symptoms in subjects affected with protein S deficiency are very similar to those with protein C deficiency. Thus, deep vein thrombosis (DVT) in the lower limbs accounts for about 90% of the episodes. Superficial thrombosis seems to be more common than for antithrombin deficient subjects.
Classification of protein S deficiency
The current subclassification of different types of protein S deficiency was recommended by the Scientific Standardization Committee (SSC) of ISTH in 1992.
| Classification SSC 1992 | Classification Comp 1990 | Protein S antigen, total | Protein S antigen, free | Protein S activity |
| Type I | Type I | Low | Low | Low |
| Type II | Type IIb | Normal | Normal | Low |
| Type III | Type IIa | Normal | Low | Low |
Type II deficiency seems to be quite rare. The Type I and Type III classification is questioned since during recent years a number of publications show that these deficiencies are present within the same families and indeed appear to be different phenotypic expressions of the same protein S genotype. Thus, in one study coexistence of both phenotypes were found in 14 of 18 families and this could be explained by the large overlap with the normal range of total protein S levels for the individuals with low free protein S antigen. These findings have been confirmed through identification of the actual mutation(s) which cause protein S deficiency in several families. Thus it was found that in one large family with no less than 122 members available for analysis, one single mutation (Gly295 Val) caused protein S deficiency in 17 family members and both phenotypes could be linked to this mutation. In all affected members, free protein S antigen was low. Total protein S antigen was low (resulting in Type I deficiency) or normal (resulting in Type III deficiency) and the difference could be explained with increasing total protein S levels with age. Later, extended studies with characterization of mutations in 9 protein S deficient families confirmed that both phenotypic expressions were found for one given mutation, the different classification being explained by not only age but also different levelsof C4BP and effects on expression levels.
The situation regarding classified Type I and Type III deficiency may still need some further clarification, though, since the consistency in the presence of a specific mutation does not seem to be universal.
Altogether, these findings strongly suggest that free rather than total protein S should be measured in the diagnosis of protein S deficiency. Restricting the analysis to free protein S still needs careful evaluation of the assay performance. This has been shown in a study on several protein S methods for detection of protein S deficiency. Another interesting finding among Type I and Type III deficiencies is the common presence of a mutation T -> C in codon 460, which results in a substitution of Ser by Pro, denoted PS Heerlen. The frequency of this allele in thrombophilia subjects is 0.7% which is not different from the 0.5%-0.8% found in the general population. Thus, PS Heerlen per se does not seem to be associated with an increased thrombotic risk. A plausible explanation for its link with, or cause of, Type III deficiency is that C4BP may bind two molecules of PS Heerlen as compared to one molecule of native protein S. Alternatively, PS Heerlen has a higher clearance rate.
The high prevalence in some families with thrombophilia might then be due to an increased risk with this allele when occurring in combination with other genetic defect(s). More knowledge will certainly be needed in this area in order to delineate the effect of this mutation.
Difficulties with protein S gene analysis
The complexity of the active PSa gene and the presence of the Protein S pseudogene PSb has delayed the genetic analysis of symptomatic protein S deficient subjects. A further complication is that no mutations at all are found in about 40% of the subjects, although all exons and flanking regions have been amplified and sequenced.
Thus, it was not until the mid-90´s that progress began. The SSC-ISTH protein S database is now rapidly growing and about 140 different mutations have been reported up to 1999.
Number of mutations found in the anticoagulant system connected with inherited thrombophilia
| Antithrombin deficiency | >79 |
| Protein C deficiency | >160 |
| Protein S deficiency | > 140 |
| APC resistance | 1 |
| Prothrombin | 1 |
| Type I deficiency results from frameshift and nonsense mutations and larger deletions and also missense mutations Type | |
Protein S deficiency in familial thrombophilia
In families with inherited thrombophilia, the prevalence of protein S deficiency is higher than in unselected thrombosis subjects. It has been increasingly clear that familial thrombophilia is often linked with more than one genetic defect, and it may well be that protein S deficiency confers a pronounced risk (6 to 10-fold increase) primarily in subjects who have one or more other inherited defects. In families where more than one hemostatic disorder has been identified, the increased risk with combined deficencies has been clearly demonstrated.
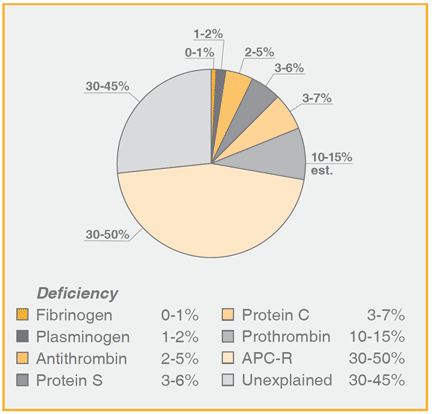
Thus in families with combined protein S deficiency and cosegregation of the factor V Leiden mutation (FV:Q506), the incidence of thrombosis increased from approximately 20% to 70% in subjects with a single defect or with both abnormalities. The high incidence of thrombosis with a single defect (about 20%) was much higher than expected for carriers of the factor V Leiden mutation, strongly indicating the presence of an additional inherited defect.
Frequency of thrombotic episodes in families deficient in antithrombin, protein S or protein C and with presence or absence of factor V:Q506
| Antithrombin 50% | FV:Q506 20% | AT + FV:Q506 91% | H.H. van Boven et al, Thromb Haemost 75, 417 – 21 (1996) |
| Protein C 31% | FV:Q506 13% | Pr C + FV:Q506 73% | B.P.C. Koeleman et al, Blood 84, 1031 – 35 (1994) |
| Protein S 19% | FV:Q506 19% | Pr S + FV:Q506 72% | B. Zöller et al, Blood 85, 3518 – 23 (1995) |
Protein S deficiency and arterial thrombosis
There are reports that protein S deficiency may be a risk factor for arterial disease in young subjects but this does not appear to be the case for the general thrombotic population.
In another study it was shown that increased thrombotic risk, both venous and arterial, was associated with the combination of homozygosity for the 4G allele in the promoter of the plasminogen activator inhibitor-1 (PAI-1) gene and protein S deficiency.
Similarly, a connection between protein S deficiency and fibrinolysis abnormality was suggested in an earlier study.
Acquired protein S deficiency
Acquired protein S deficiency has been demonstrated in several disease states, such as in liver disease, disseminated intravascular coagulation and in subjects with chronic inflammatory bowel disease.
An acquired deficiency state may occur in subjects with antiphospholipid antibody syndrome.
The mechanism behind this is not clear but one interesting study has shown that b2-Glycoprotein I interferes with the binding of protein S to C4BP and hence antibodies against b2-Glycoprotein I may result in a decreased level of free protein S due to its excessive binding to C4BP.
Clinical features and management
Management of thrombosis subjects with protein S deficiency or another inherited coagulation disorder generally does not differ from other thrombotic subjects. For both symptoms and management, see Lane et al 1996 for a review.
Elevated protein S levels and ischemic heart disease
Interestingly, a study on 150 individuals has shown an association between free protein S antigen and, to a lesser extent, total protein S with cholesterol and triglycerides, using a Laurell electroimmunoassay for total and free protein S.
There was a rise in free protein S antigen with 30% between the 5th and 95th percentiles of triglyceride concentrations and a 10% rise of total protein S between these percentiles for cholesterol.
For protein C an increase of 25% was noticed for both lipids. These results and the difference between men/postmenopausal women and premenopausal women as well as the lower levels obtained in women using oral contraceptives indicate that great caution should be used when a normal range is to be established.
Findings in agreement with this were indeed noticed in a recent prospective study where an elevation in free protein S was associated with an increased risk of ischemic heart disease and thereby paralleling the increase in known risk factors for this disease.
Protein S Assays Methods
The development of suitable assays for the measurement of protein S in plasma is hampered by the fact that PS circulates in the plasma both as free and in complex with C4BP. Basically three types of assays can be recognized:
- Immunological assays for total protein S
- Immunological assays for free protein S
- Functional assays for protein S
Total Protein S antigen
Numerous immunological assays for the measurement of total protein S in plasma have been described and they include: Laurell-type of assay, enzyme linked immunosorbent assay (ELISA), immunoradiometric assays (IRMA), and radioimmunoassay (RIA).
The selection of a suitable assay is made on the basis of sensitivity and specificity of the different assays available on the market. Important criteria for selection of a total protein S antigen assay should be a demonstration that free and complexed protein S are recognized to the same extent by the selected antibody and that the result is not influenced by variations of the C4BP-protein S plasma concentration. Alternatively, high plasma dilutions and long incubation times are required to favor a complete dissociation of C4BP-protein S complexes.
Free Protein S
Free Protein S antigen
As mentioned in the previous paragraphs, protein S and C4BP form a complex under physiological conditions. In citrated plasma about 40% of the total protein S is free while the remaining 60% is bound to C4BP. The amount of free protein S corresponds to the molar excess over C4BP in plasma. Since only the free part of total protein S antigen has an APC-cofactor activity, it is of importance to know the concentration of the free protein S.
The current indication is that the assays for free protein S have higher sensitivity and specificity for the genetic defects causing protein S deficiencies than the assays for total protein S. This is because there is a great overlap in the total protein S levels between normal and those with the genetic defects. Free protein S antigen can be quantitatively determined after first precipitating the C4BP-protein S complex with polyethylene glycol and determining free protein S in the supernatant after centrifugation. The measurement of free protein S antigen in PEG supernatant sometimes creates a problem when a protein S antigen test, that is not really specific for total protein S, is used. For this reason most authors express the concentration of free protein S antigen as a percentage of the free protein S antigen present in normal plasma. ELISA methods have also been developed in which the PEG precipitation step is omitted and where free protein S antigen is directly determined with specific monoclonal antibodies.
Protein S activity assays
Although the anticoagulant mechanism of protein S in vivo has not been elucidated, there should be no doubt of its importance as an anticoagulant protein. This is obvious, as mentioned above, from the severe massive thrombosis which occur in neonates who have homozygous protein S deficiency. A strong support that only free protein S has anticoagulant activity was shown in a study in which addition of a stoichiometric excess of C4BP over protein S before infusion of E.Coli resulted in lethal coagulopathy. In contrast, supplementation with a slight excess of protein S together with C4BP prevented a fatal outcome. Thus, determination of free protein S antigen (see above) or activity is of importance. In functional protein S activity assays, which are in routine use, the effect of free protein S as a cofactor to APC is determined. These assays are predominantly coagulometric and measure the prolongation of the clotting time due to free protein S activity in the degradation of factor Va and factor VIIIa by APC Free protein S also exerts an APC-independent anticoagulant activity through direct binding to factor Va, factor Xa and factor VIII and these interactions appear to be phospholipid-dependent.
An assay of the APC-independent anticoagulant activity of protein S with improved resolution has been developed in which the clotting time is determined in the presence and absence of a polyclonal protein S antibody.
However, no assays specific for this APC-independent activity of protein S seem to be in routine use so far.
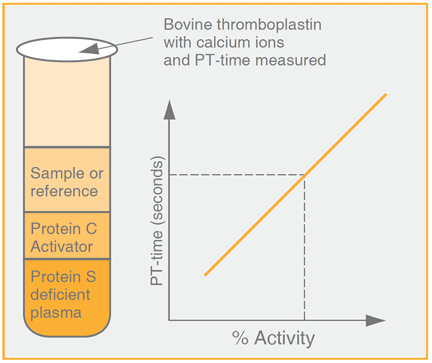
APC cofactor methods for free protein S activity
Initially, methods were developed which used undiluted test plasma. However, this approach turned out to be inadequate due to the great influence of prothrombin levels, with increasing amounts of prothrombin resulting in decreasing clotting times. Later methods all utilize an excess of protein S deficient plasma, thereby keeping the amount of prothrombin essentially constant.
These methods can be categorized as follows:
- Activated partial thromboplastin time (APTT) method variants, using either addition of exogenous APC or addition of Protac® C for activation of protein C present in the test plasma and in the protein S deficient plasma. In these methods, the cofactor activity of protein S in the APC-dependent degradation of both factor Va and factor VIIIa is analysed. By adding factor Va, a greater sensitivity is obtained, which allows for the use of more diluted test plasma and a seemingly improved specificity. Depending on the actual assay system used, a resolution of 15-60 seconds is obtained between 0 and 100% protein S.
- Prothrombin time (PT) method. The cofactor activity of protein S is confined to the APC-dependent degradation of factor Va. Originally, a method was developed for characterization of purified protein S which was later followed by a functional test for determination of protein S in plasma. In the latter method, Protac C is used for activation of protein C contained in protein S deficient plasma prior to addition of diluted test plasma. This method was designed for easy application on automated analyzers. shows the schematic design of this method. A resolution of 40-50 seconds is obtained between 0 and 100% protein S.
- Factor Xa-based methods. In these methods, coagulation is triggered by factor Xa in the presence of calcium ions and phospholipids. Originally undiluted plasma was used. Due to the drawback mentioned above, this was later replaced by more sensitive methods allowing dilution of test plasma and providing close to 100 seconds resolution between 0 and 100% protein S. In one variant of the method, free protein S in the test plasma is first adsorbed on an insolubilized monoclonal protein S antibody. Factor Xa has also been used as trigger in a system utilizing purified components. It should also be mentioned that although preliminary and promising results have been presented with chromogenic protein S methods, no such methods are currently used in routine settings.
Interference in protein S clotting assays by APC resistance due to the FV:Q506 (FV Leiden) mutation
In general, protein S clotting assays for determination of free protein S activity do not show an impressive comparability on analysis of subject plasmas, whereas more consistent results are obtained on analysis of normal plasmas.
The lack of a proper agreement in subject plasmas may in part be due to various influences on the effects of dysfunctional protein S and also to acquired disorders.
As far as APTT-based methods are concerned, it has been shown that different APTT reagents show pronounced differences in the sensitivity towards the APC cofactor activity of protein S.
However, by far the most serious interference is caused by APC resistance due to the FV:Q506 mutation. This mutation causes an impairment in the degradation of factor Va by APC and has been shown to result in misclassification of subject plasmas as Type II protein S deficiency. By taking care to exclude carriers of the factor V mutation as donors in preparation of protein S deficient plasma such interference seem to be reduced. Increased experience from analysis of subjects homozygous for the factor V mutation should be gained.