Technochrom FVIII:C 2G
$0.00
Technochrom® FVIII:C 2G is a chromogenic assay for the in vitro measurement of Factor VIII activity in citrated plasma. This Factor VIII test kit has two measuring ranges and is fully automatable on the Ceveron® 100 Series.
Measurement Principle
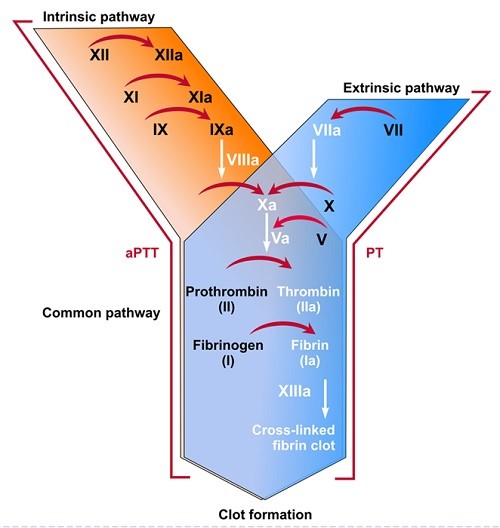
Factor VIII (FVIII), when activated, functions as a cofactor for activation of FX by activated FIX (FIXa) in the complex interplay of blood coagulation biochemistry, ultimately leading to fibrin formation that strengthens and stabilizes a blood clot.
Technochrom FVIII:C 2G is a two-stage chromogenic assay of FVIII cofactor activity. In the first stage, diluted plasma containing the FVIII to be measured is activated with thrombin (FIIa), and then when it also reacts with exogenous FIXa, FX, phospholipids, and Ca2+, it forms the tenase (FVIIIa-FIXa-FX) complex. The product of tenase is activated FX (FXa), where the amount generated is directly proportional to the FVIII level in the sample. In the second stage, FXa cleaves the chromogenic substrate to release a colored product, para-nitroaniline, the intensity of which is proportional to the FXa, and hence, proportional to the FVIII cofactor activity. Absorbance is measured at 405 nm.
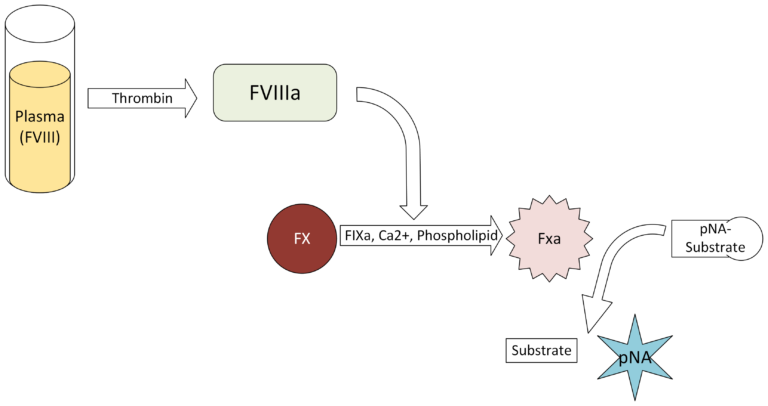
Kit Composition
- Substrate 2×10 mL lyophilized chromogenic substrate (Z-D-Arg-Gly-Arg-pNA), a thrombin inhibitor, TRIS, EDTA, and Sodium Chloride. Add 10 mL purified water and incubate for 15 minutes to reconstitute.
- Reagent A 2×2 mL lyophilized reagent containing phospholipids, BSA, TRIS, and a heparin neutralizer. Add 2 mL purified water and incubate for 15 minutes to reconstitute.
- Reagent B 2×2 mL lyophilized reagent containing bovine Factor X, human Factors IXa and FIIa, BSA, TRIA, sodium chloride, calcium chloride, and a fibrin polymerization inhibitor. Add 2 mL of purified water and incubate 15 minutes to reconstitute.
Background
Coagulation Factor VIII (FVIII, F8), also known as anti-hemophilic factor, is a crucial protein in hemostasis and coagulation, the process that stops bleeding by forming blood clots. Understanding the structure, function, and genetic aspects of Factor VIII is essential to fully appreciating its importance to human physiology. Factor VIII is a large glycoprotein produced primary in the sinusoidal cells of the liver. It circulates in the blood in an inactive form bound to von Willebrand Factor (vWF). vWF is a carrier protein that stabilizes Factor VIII, preventing premature degradation. Upon vascular injury, Factor VIII is activated and separates from vWF. The active form of Factor VIII, FVIIIa, acts as a cofactor for Factor IXa in the conversion of Factor X to Factor Xa, the crucial step in the coagulation cascade. This cascade is a series of enzymatic reactions that culminates in the formation of a fibrin rich blood clot; with Factor VIII enhancing the efficiency of Factor IXa, accelerating the production of FXa and ultimately leading for the formation of a stable fibrin clot.
The gene that encodes Factor VIII—known as F8—is located on the X chromosome (Xq28). This explains why hemophilia A, the disorder caused by a deficiency of Factor VIII, as an X-linked recessive condition primarily affects males. While males only have a single copy of the X chromosome, females with a defective F8 gene are protected by their second, normal, copy of the F8 gene, making them carriers for hemophilia. Mutations in the F8 gene can lead to varying levels of FVIII deficiency, resulting in mild, moderate, or severe forms of hemophilia A. These mutations can range from small base replacements to inversions, insertions, and even large deletions. Each mutation affects the proper function of the protein differently.
Hemophilia A is the most well-known condition associated with Factor VIII. This disorder is characterized by episodes of spontaneous bleeding, prolonged bleeding after injury, and hemorrhages of both the joints and muscles. The severity of hemophilia A correlates with the amount of FVIII activity in the blood. A mild case of hemophilia will retain 5-40% of normal activity while a severe case will retain less than 1% of normal FVIII activity. In individuals affected by FVIII deficiency, levels of FVIII activity can be replaced by the injection of plasma donor derived or recombinantly synthesized Factor VIII. More recently, gene therapies have started to be used to increase Factor VIII levels in a more sustained and longer-term manner than strict replacement.
Factor VIII circulates in plasma bound to von Willebrand factor (VWF), which protects FVIII from premature clearance. Hereditary deficiency of FVIII causes Hemophilia A (HA), an X-linked bleeding disorder characterized as mild, moderate, or severe, based on degree of FVIII reduction. Those with HA can develop inhibitory antibodies to FVIII, and acquired HA can arise from development of autoantibodies to FVIII. Both FVIII and VWF are acute phase reactants, and elevated FVIII is associated with thrombosis.
Research into the structure, function, and regulation of Factor VIII continues to advance our understanding of this crucial gene and protein. Recent efforts have been made to develop longer lasting FVIII products and to explore gene editing technologies for correcting F8 mutations at the DNA level.