Chromogenix S-2403™
$0.00
Chromogenix S-2403™ is a chromogenic substrate for plasmin and streptokinase-activated plasminogen.
Each vial contains the chromogenic substrate S-2403™ 25 mg and mannitol 60 mg as a bulking agent.
Stability
- Substance: Stable until expiry date if stored at 2-8°C. Avoid exposure to light. The substance is hygroscopic and should be stored dry.
- Solution: 7.5 mmol/L in H2O is stable for at least 6 months at 2 to 8°C.
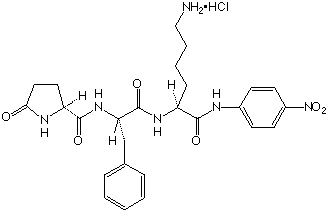
| Chemical name | L-Pyroglutamyl-L-Phenylalanyl-L-Lysine-p-Nitroaniline hydrochloride. |
| Mol. wt | 561.0 |
The majority of the Chromogenix substrate library has an Arginine (Arg or R) group at the P1 position (the amino acid position that occurs at the preferred cleavage site). Why is this?
The Chromogenix line is geared toward the proteins involved in hemostasis. These are a group of proteolytic enzymes that comprise the serine proteases, which cleave mainly at the C-terminal side of the basic amino acids arginine or lysine. The peptides at the P2, P3, and P4 positions contribute to the substrate’s specificity. Note that the substrates for plasmin cleave at a lysine group. Other protease groups are aspartic proteases (like pepsin), metallo proteases, and cysteine proteases (which include caspases, with an asp cleavage site).
There are a few different substrates that are hydrolyzed by plasmin. If I want to use as short incubation times as possible, and need a selective substrate for plasmin, which should I choose?
The substrates for plasmin include S-2251™, S-2302™, and S-2403™. While S-2251™ is a popular and suitable substrate for detection of plasmin, S-2403™ has a higher kcat/km value. It is a faster substrate, and incubation times can be shorter. S-2403™ is therefore the substrate of choice for this situation.
Which substrate is best suited for measuring two-chain tPA, and why?
S-2765™, S-2366™, and S-2288™, S-2403™ are suitable for single-chain tPA, and S-2251™ was used in the discontinued Chromogenix Coaset® tPA kit, but there are no substrates specifically for two-chain tPA. A paper by Verheijen et al. (Thromb Res, 1985; 39: 281 – 288), however, describes a method comparing the direct amidolytic activity of tPA on S-2366™ and the plasminogen activating activity. Also, the substrate S-2288™ is suitable for measuring double-chain tPA because it has a slightly higher sensitivity than S-2366™. S-2288™ should be used with purified systems, though, since this substrate is sensitive for several proteases.
What if the reconstituted substrate has some precipitate or is cloudy?
The substrate solution is usually prepared with sterile water, but sometimes they may not dissolve properly. Sonication may help, or substrates with low solubility in water can be dissolved in DMSO, then diluted in water. The final DMSO concentration should preferably not exceed 10% in the reaction mixture. It should be noted that stability in DMSO is decreased, as it also is with alkaline buffers.
Why is pNA the leaving group on all of the Chromogenix substrates?
A good chromophore must be readily cleaved by and dissociated from the enzyme. The color must be strong to allow detection of low enzyme activities, but should not interfere with the color of other reactants or impurities in the reaction mixture. It should be water soluble and have low toxicity. The chromophore para-nitroaniline (pNA) fulfills most of these requirements. It is therefore the most common choice of chromophore.
Why are the reactions measured at 405 nm?
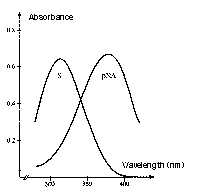
The absorption intensity is expressed by the Beer-Lambert law, A = e x c x l, where A is absorbance, c is molar concentration, l is the path length of sample cell (usually 1 cm), and e is the extinction or molar absorptivity coefficient. The absorption spectrum of the substrate versus the chromophore, pNA (the chromogenic leaving group) is the reason for reading the reactions at 405 nm. The absorbance maximum of the unhydrolyzed, intact substrate is 316 nm and 380 nm for pNA. Although the difference between substrate and product is maximal at 385 nm, at 405 nm, there is less background reading, and the absorbance of the substrate is still less than 1% of that of an equimolar amount of pNA.
What aspects must a scientist consider when choosing the best chromogenic substrate?
Synthetic substrates are very sensitive; they can detect very low enzyme activities and are often more sensitive than a corresponding natural substrate. On the other hand, they can be less selective, or, have less discrimination in their reactivities toward related enzymes compared to the natural substrate. There are steps a scientist can take to maximize sensitivity and specificity. If the specificity of the enzymatic activity to be measured is known then a substrate selectivity table which shows the cross-reactivity of the substrates with different enzymes, and the kinetic data, such as that provided in the Chromogenix catalog, can be helpful. If the specificity of the enzyme is unknown, a screening procedure can be applied. This involves comparing the rate of hydrolysis obtained with different substrates. The presence of contaminating enzymes must also be taken into account. To eliminate interference, an inhibitor can be introduced, the sample can be further diluted, or conditions can be found where the relative activities are optimized. For instance, S-2222™ is selective for FXa, but also for trypsin. If a researcher wants to measure FXa, s/he can add an inhibitor to trypsin, such as soybean trypsin inhibitor. Temperature, pH, buffers, and ionic strength can all affect the rate of hydrolysis and must be considered. Substrate concentration is also important, and a concentration of 2 x Km is usually appropriate. A good substrate has a low Km, meaning maximum reaction velocity is achieved at a low substrate concentration. In other words, the enzyme has a high affinity for the substrate. A high Kcat is also desired, which means the enzyme has a high turnover rate with the substrate (fast reaction).
Define a katal.
One katal (kat) is the amount of enzyme that converts one mole of substrate per second. Activated enzymes from Chromogenix such as FXa and thrombin are measured in nkat. 1 nkat = 1 x 10-9 mol of product released per second.
What is a chromogenic substrate composed of?
Chromogenic substrates are peptides that react with proteolytic enzymes under the formation of color. Chromogenic substrates are made up of a protecting group, amino acid residue(s), side-chain modification if applicable, and the chromophore. The stereochemistry of some substrates may be designated. For example, in the Chromogenix substrate S-2222™, the protecting group is a benzoyl group, the amino acid residue is Ile (isoleucine – a non-polar hydrophobic amino acid), the side chain modification is Glu(g-OR)- where R is 50% H (hydrogen) and CH3 (methyl group). The P2 and P1 amino acid residues are Gly and Arg, respectively, and the chromophore is pNA (para-nitroaniline).
Define an enzyme and a substrate.
Enzymes are proteins that catalyze most of the chemical reactions that take place in the body. The chemical compound upon which the enzyme exerts its catalytic activity is called a substrate. Proteolytic enzymes degrade their substrates, proteins and peptides, by hydrolyzing one or more peptide bond(s). For information on enzyme kinetics, see the Chromogenix Catalog or contact Diapharma Group, Inc. at info@diapharma.com.
What is a peptide? What is the difference between a tripeptide and a tetrapeptide? How are amino acids linked to form peptides?
A peptide is the name assigned to short polymers of amino acids. They are classified by the number of amino acid units in the chain, called amino acid residues. Tripeptides have three amino acid residues while tetrapeptides have four. A polypeptide is formed when the chain of amino acid residues exceeds several dozen in length. A protein is a molecule composed of one or more polypeptide chains.
Proteins are unbranched polymers of amino acids linked head to tail from carboxyl group to amino group, through formation of covalent peptide bonds. The peptide backbone of a protein consists of the repeated sequence.
-N-Ca– C, where N represents the amide nitrogen, Ca represents the a-carbon atom of an amino acid in the polymer chain, and the final C is the carbonyl carbon of the amino acid. This C is in turn linked to the amide N of the next amino acid, and so on down the line. The unbranched polypeptide chain has two ends, an amino-terminal or N-terminal end and a carboxyl-terminal or C-terminal end.
What substrate research methods are available?
- Chromogenix Chromogenic Substrate S-2222 Trypsin
- Chromogenix Chromogenic Substrate S-2222 Tissue Factor Pathway Inhibitor, type – I (TFPI)
- Chromogenix Chromogenic Substrate S-2238 Antithrombin
- Chromogenix Chromogenic Substrate S-2238 Antithrombin (anti-FIIa)
- Chromogenix Chromogenic Substrate S-2288 Proteolytic activity
- Chromogenix Chromogenic Substrate S-2238 Thrombin
- Chromogenix Chromogenic Substrate S-2228 Tissue Plasminogen Activator (t-PA)
- Chromogenix Chromogenic Substrate S-2251 Plasmin
- Chromogenix Chromogenic Substrate S-2302 Kallikrein
- Chromogenix Chromogenic Substrate S-2302 Kallikrein Inhibitor
- Chromogenix Chromogenic Substrate S-2302 Prekallikrein
- Chromogenix Chromogenic Substrate S-2366 Hirudin
- Chromogenix Chromogenic Substrate S-2765 Factor X
- Diapharma Chromogenic Substrate CS GK (substitute for discontinued Chromogenix S-2266) Kallikrein in urine
- Diapharma Chromogenic Substrate CS UK (substitute for discontinued Chromogenix S-2444) Urokinase
- Diapharma Chromogenic Substrate CS PSA (KLK3) (substitute for discontinued Chromogenix S-2586) Chymotrypsin
Historically, the development of a new chromogenic substrate for a specific protease has always been accompanied by the release of method sheets where the application and the methodology for a particular use were described in detail. Some methods are simple chromogenic assays where a buffer and the substrate are the only reagents to be used (i.e. proteolytic activity). Other methods instead require the use of additional compounds, which are or have been commercially available from Chromogenix or elsewhere, and consist of more reaction steps. These protocols were validated in laboratories according to the equipment and the reagents available at the time. In several cases they have been adopted in research, quality control, or routine laboratories and some of them later became Chromogenix kits now present in our product range.
During the last 20 years, the so-called Method Sheets have been taken as the starting point by several scientists, for the development of assays for particular applications, or studies. In some cases the experimental conditions have been changed according to the particular need of the investigation being done. These methods are now presented in a different form: “Research Methods”. The intention here is to provide assay protocols that are not available as kits, but complementary to our product range. In the following list, you can find the methods developed by Chromogenix for several assays. They have to be considered as general guidelines or basic tools for the development of your own assays, some of them requiring a validation within your laboratory with respect to the reagents and equipment used.
For each method, you can find an updated Bibliography with references, where the method has been used, like as originally described or with modifications. This information should facilitate and accelerate the development of the best test protocol. If you do not have that specific journal issue in your laboratory, you can visit MEDLINE. You can search for particular articles, print the abstract and order the original copy through LOANSOME DOC (and receive the document through your local library). At the same time, on our web site you have the possibility to be updated on the new products from Chromogenix.
Theoretical basis for calculation
The hydrolysis of the chromogenic peptide substrate by the proteolytic enzyme follows in general the Michaelis-Menten kinetics. This means that, if the substrate is present at a sufficiently high concentration or if a comparatively small fraction of the substrate is hydrolized, the rate of product (color) formation is proportional to the activity of the enzyme. The rate of pNA formation, i.e. the increase in absorbance per second, is measured photometrically at 405 nm. At this wavelength the extinction coefficient of pNA is
9600 mol -1 • l • cm -1
The enzymatic activity can be quantified in two ways:
- By comparing the activity of an enzyme with that of a standard preparation, which is defined in terms of a specified number of units set by an international or national authority or society (WHO, NIH etc.), or by the activity present in 1 ml of activated pooled normal plasma (Plasma Equivalent Unit = PEU). The standardization is performed by using at standard curve obtained with at least five different concentrations, each performed in duplicate. The standard material must be of the same kind and of the same quality as the sample which is to be measured. This may be still more important for a secondary or domestic standard.
- By measuring the amount (mol) of substrate split, or rather product formed per unit time (absolute activity).
One unit of enzymatic activity, katal (kat) is defined as the amount of activity that converts one mole of substrate per second under standardized conditions. Such conditions as type of substrate, substrate concentration, buffer, pH, ionic strength and temperature should be given along with unit.
Thus, 1 nkat gives a conversion rate of:
1 x 10-9 mol/sec = 60 x 10-9 mol/min
If the total (measuring) volume used is V (ml), the increase in concentration per minute caused by 1 nkat is
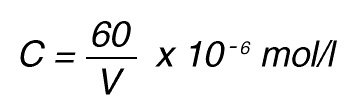
If the absorbance is measured at 405 nm, in a 1 cm cuvette the difference in extinction coefficient is
e = 9600 mol-1 • l
The increase in absorbance/min can then be calculated by using Lambert-Beer’s law:
A = e x C
Thus, 1 nkat gives:
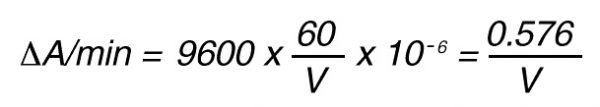
or
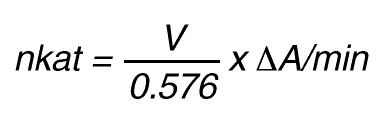
By using a sample volume v (ml):
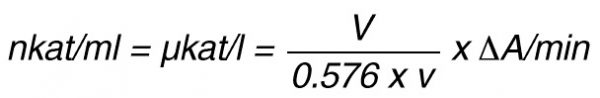
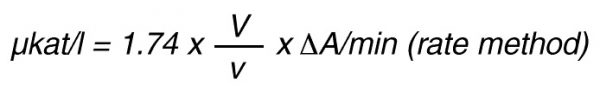
For the end-point method, the incubation time t (min) with substrate is taken into account by the following formula:
According to nomenclature, one unit (U) is the amount of enzyme activity that converts one mol of substrate per minute under standardized conditions. By using the above formulas the units are:
Protein concentrations in plasma
| Component | Molecular Weight kDa | Plasma Concentration mg/l | Plasma Concentration μmol/l |
|---|---|---|---|
| Fibrinogen | 330 | 3000 | 9 |
| Prothrombin | 72 | 150 | 2 |
| Factor V | 330 | 20 | 0.05 |
| Factor VII | 50 | 0.5 | 0.01 |
| Factor VIII | 330 | 0.1 | 0.0003 |
| Factor IX | 56 | 5 | 0.09 |
| Factor X | 59 | 8 | 0.13 |
| Factor XI | 160 | 5 | 0.03 |
| Factor XII | 80 | 30 | 0.4 |
| Factor XIII | 320 | 10 | 0.03 |
| Protein C | 62 | 4 | 0.06 |
| Protein S | 70 | 10 (free) | 0.14 |
| Protein Z | 62 | 2 | 0.03 |
| Prekallikrein | 86 | 50 | 0.6 |
| HMW kininogen | 120 | 70 | 0.6 |
| Fibronectin | 450 | 300 | 0.7 |
| Plasminogen | 92 | 200 | 2 |
| t-PA | 60 | 0.005 | 0.0001 |
| Urokinase | 53 | 0.004 | 0.0001 |
| Antithrombin | 58 | 145 | 2.5 |
| Heparin Cofactor II | 66 | 80 | 1.2 |
| Plasmin Inhibitor | 63 | 60 | 1 |
| Protein C Inhibitor | 57 | 4 | 0.07 |
| α2-Macroglobulin | 725 | 2000 | 3 |
Measurement Principle
The method for the determination of activity is based on the difference in absorbance (optical density) between the pNA formed and the original substrate. The rate of pNA formation, i.e. the increase in absorbance per second at 405 nm, is proportional to the enzymatic activity and is conveniently determined with a photometer.