Detecting Hemoglobin Variants during Sickle Cell Disease Research: Which Method is Best?
Posted on: March 15, 2021
– Contributed by Abi Kasberg, PhD
Hemoglobins are protein molecules found in red blood cells (RBCs) that functionally transport oxygen from the lungs to the cells of the body and to shuttle carbon dioxide back to the lungs. Hemoglobins also play important roles in maintaining RBC shape by dynamically changing conformation through binding oxygen. Hemoglobin is composed of four globin subunits, the most common being the alpha, beta, and gamma chains. Each globin subunit contains a heme group that carries iron that can bind one molecule of oxygen each. Fetal hemoglobin (HbF) is produced in fetuses and newborns and is made up of two alpha and two gamma-globin chains (Figure 1). HbF makes up roughly 85% of hemoglobin at birth, and then falls to less than 1% of total hemoglobin by 1 year of age. Shortly after birth, beta chains gradually replace gamma chains, resulting in adult hemoglobin (HbA) production (Figure 1). HbA is normally composed of two alpha and two beta-globin chains. This transition from HbF to HbA is mediated by the BCL11A gene. The BCL11A transcription factor silences the expression of fetal gamma-globin chains shortly after birth.
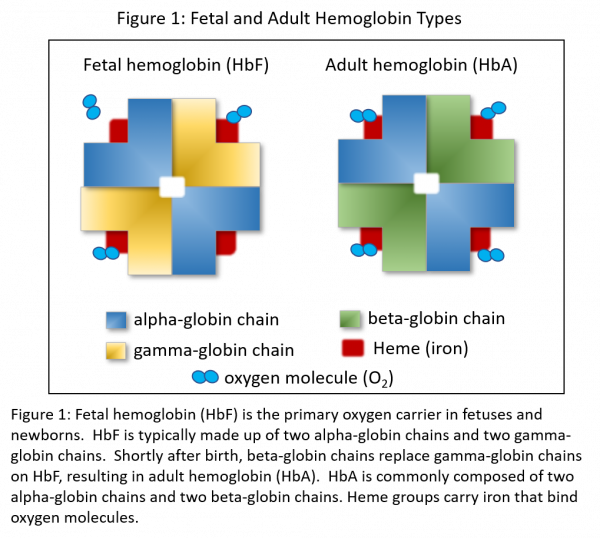
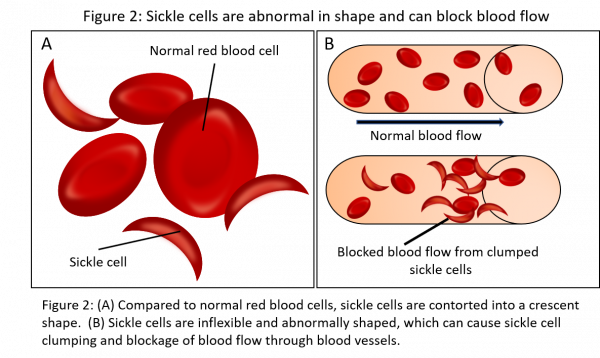
Sickle cell disease (SCD), also called sickle cell anemia, is an inherited condition in which RBCs contort into a crescent shape due to an abnormal hemoglobin variant (HbS). These abnormal RBCs break down and experience accelerated apoptosis resulting in a truncated life span. This drives a shortage of healthy RBCs that can lead to anemia, which causes fatigue and shortness of breath. Due to the abnormal sickle shape and inflexibility of RBCs in SCD, blood vessels and blood flow can become blocked from clumped sickle cells (Figure 2). This can drive episodes of pain, pulmonary hypertension, organ damage, and heart failure.
SCD is caused by a genetic mutation in the HBB gene that expresses the beta-globin chain of adult hemoglobin. This atypical beta-globin chain is called sickle hemoglobin (HbS). Because HbF does not contain the beta-globin chain, HbF cannot induce RBC sickling. In fact, infants who carry the HbS variant and have SCD display few symptoms with RBCs and rarely sickle. High levels of HbF in fetuses and newborns inherently protects from sickling outcomes, even in SCD carriers. This is an important and fascinating phenomenon to consider, particularly while developing SCD therapeutics.
Recent Gene Therapies to Treat Sickle Cell Disease
Because SCD is a heritable disease, gene therapy strategies are being developed as treatments for SCD. Popular strategies include globin gene addition, gene editing to fix the HBB mutation, or genetic maneuvers to boost HbF production. The promotion of HbF production is achieved through blocking the expression of the HbF inhibitor, BCL11A, in RBCs. BCL11A acts as a gatekeeper, or switch, to regulate HbF levels. Therefore, blocking BCL11A expression will enable an increase of HbF, the hemoglobin which does not sickle. The ultimate goal of this strategy is that HbF will become the principal hemoglobin through hemoglobin switching. This enables an environment where HbF is available to compensate for the SCD HbS variant, and hence rid the individual of sickled hemoglobin and SCD symptoms.
Two recent trials have reported results using this gene therapy strategy to treat SCD. A cutting-edge gene-editing trial used CRISPR-CAS9 genome editing technology to target BCL11A (Frangoul et al. 2021). A separate study used a lentiviral vector to encode shRNA that targets and silences BCL11A mRNA (Esrick et al. 2021). In both trials, blood stem cells were removed from the bone marrow of SCD individuals. Hematopoietic stem cells were modified in a laboratory and then infused back into the subjects. Preliminary results from both studies were promising and reported that subjects experienced a decrease in SCD symptoms following gene therapy treatments.
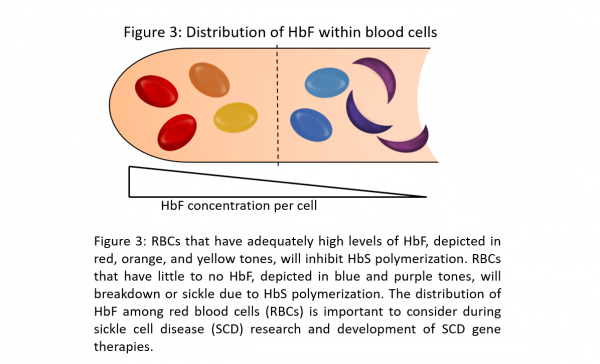
A critical component of SCD gene therapeutic research is evaluating HbF, HbA, and HbS hemoglobin concentration and distribution. Blood concentration values measure the number of cells that carry a particular hemoglobin, but blood concentration values do not measure the quantity of hemoglobin molecules per cell. Similarly, total hemoglobin measurements within a hemolysate sample provide a limited snapshot of an otherwise dynamic hemoglobinopathy disease. To provide a thorough analysis, it is essential to evaluate hemoglobin distribution within a sample. This is because high levels of the non-sickling HbF can be associated with severe SCD due to an uneven distribution of HbF molecules among blood cells (Steinberg et al 2014). Despite high blood concentrations and total amounts of HbF, individual RBCs may be void of HbF and are unable to overcome HbS polymerization, resulting in RBC sickling and destruction (Akinsheye et al 2011) (Figure 3). Therefore, it is critical for the development of SCD therapeutics to evaluate the distribution of HbF and HbS among RBCs in addition to measuring total hemoglobin levels.
Methods for Detecting Hemoglobin Variants
There are several methods available for detecting HbS, HbF, and other hemoglobin variants. The most common method to quantify hemoglobin in SCD is through high-performance liquid chromatography (HPLC). HPLC separates and quantifies hemoglobin fractions, based on known retention times required for gradient elution (Nair 2018). Each hemoglobin fraction, based on the predetermined retention time, is calculated, and plotted on a chromatogram in the form of graphical peaks. Therefore, it is possible—and even likely—that there are wide ranges of specificity within retention times, such that hemoglobin variants will elute together in the same detection window having overlapping peaks. HPLC evaluation of hemoglobin variants are dependent on peak shape, position, and retention time, which have limitations on specificity (Nair 2018). Indeed, HbS can elute in the same location as HbA in some fractions by HPLC, as well as with other variants, such as HbC (Keren et al 2008).
Capillary electrophoresis (CE) separates hemoglobin variants through an electrolyte solution under a generated electroosmotic flow. CE is an alternative to HPLC that requires smaller sample size and has shorter migration time frames for analyte separation. CE works best to analyze peptides that differ in net charge and is dependent on factors such as solvent pH and ionic strength. In some samples, hemoglobin variants will not separate, such as HbC from HbA2. CE outputs are easier to interpret than HPLC outputs if the sample contains HbA. When HbA is absent from a sample, an additional analysis must be done to confirm the identity of the hemoglobin (Keren et al 2008).
Flow Cytometry is an established method that provides detailed evaluations of cell populations based on cell type, quantities, morphologies, and protein markers. Flow cytometers operate by running single cell suspensions past a laser beam where scattered fluorescent light is used to count, sort, and characterize each cell. For use with SCD research, flow cytometry is a useful tool for quantifying the number of cells that carry a specific hemoglobin of interest using hemoglobin-specific antibodies. This is a valuable tool for determining the distribution of hemoglobin types within the total number of cells in the sample. Detecting HbS by flow cytometry can be more precise than other techniques and does not need to account for interference from other hemoglobin variants with overlapping peaks, like HbE.
Mass Spectrometry is a technique that can identify hemoglobin variant subunits and provides detailed data regarding the relative abundance of each hemoglobin chain within a sample. Mass spectrometry is also capable of identifying mutation sites on hemoglobin subunits and is sensitive to subtle variant differences that are undetectable using HPLC and CE conventional methods. Despite the informative attributes of mass spectrometry, the execution of mass spectrometry is technically and analytically challenging and the data output of mass spectrometry is not easy to interpret.
Single cell analysis can detect precise concentrations of hemoglobin variants within a single cell. However, this strategy is not widely used in clinical research applications. Single cell hemoglobin data can be difficult to interpret due to the dynamic changes of hemoglobin subunit concentrations during RBC maturation.
It is important to note that individuals with SCD have unique distributions of HbF per RBC despite total HbF levels (Steinberg et al 2014). Therefore, methods that measure the distribution of hemoglobin chains and variants within a sample, such as flow cytometry, are powerful tools to utilize during hemoglobinopathy research and the development of SCD gene therapeutics.
Table 1: Methods for Determining Hemoglobin Variants
| Method | Assay Strategy | Variant Specificity | Total Hemoglobin Levels | Hemoglobin Distribution |
| Capillary Electrophoresis | Separates analytes via electroosmotic flow | Limited hemoglobin variant separation | ✔ | —- |
| Flow Cytometry | Quantifies and can sort cells via fluorescent light scatter |
✔ (antibody dependent) |
✔ | ✔ |
| HPLC | Separates fractions through a column based on known retention times | Limitations due to overlapping peaks and retention times | ✔ | —- |
| Mass Spectrometry | Converts molecules to ions and then measures mass-to-charge ratios for the calculation of molecular weight | ✔ | ✔ | —- |
| Single Cell Analysis | Determination of hemoglobin variants within a single cell | ✔ |
✔ (within single cell) |
—- |
Further Reading
Akinsheye I, Alsultan A, et al. Fetal hemoglobin in sickle cell anemia. Blood. 2011 Jul 7; 118(1): 19–27.
Esrick EB, Lehmann LE, et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. N Engl J Med 2021; 384:205-215.
Frangoul H, Altshuler D, et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N Engl J Med 2021; 384:252-260
Keren DF, Hedstrom D, et al. Comparison of Sebia Capillarys Capillary Electrophoresis With the Primus High-Pressure Liquid Chromatography in the Evaluation of Hemoglobinopathies. Am J Clin Pathol 2008;130:824-831
Nair SB. Potential Pithfalls in Using HPLC and its Interpretation in Diagnosing HbS. J Rare Dis Res Treat. (2018) 3(3): 9-12
Steinberg MH, Chui DHK, et al. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood (2014) 123 (4): 481–485.
ML-00-00774 Rev01