Factor VIII is a non-enzymatic plasma protein that is essential for normal blood coagulation. The deficiency of factor VIII activity in humans is associated with a congenital bleeding disorder, called hemophilia A, which affects about 1 in 5000 males. Hemophilia A patients are treated with factor VIII concentrate for maintenance of normal hemostasis but prophylactic treatment is not in general use worldwide. During later years recombinant factor VIII has been approved for therapeutic use, which minimizes the risk of viral transmission the hemophiliacs could contract from blood transfusion products as part of the previous standard of care. In contrast to a deficiency, there is growing evidence that elevated factor VIII activity is a risk factor for thrombosis. Hence, factor VIII levels are of importance to measure not only for diagnosing and monitoring hemophilia A but also for thrombophilia investigations or research. With the advent of chromogenic substrate technology, accurate and sensitive methods are available for quality control and for the clinical coagulation laboratory. It is the purpose of this monograph to present an overview of biochemical and clinical data on factor VIII and to provide comprehensive information on methodological aspects and on the use of the Chromogenix Coamatic® and Coatest® Factor VIII kits.
Introduction and History
Hemostasis is the collective name of all the processes which allows uninterrupted blood flow and which, on demand, has an immediate and considerable capacity to stop leakage of blood from the blood organ and to dissolve small thrombi, should these events occur. Maintenance of normal hemostasis is therefore, not surprisingly, a complex and delicate balance between procoagulant and anticoagulant events and their regulation.
The bleeding disorder hemophilia A, with its easily recognized and dramatic symptoms already in childhood, has been known among mankind for a very long time.
Most people may be familiar with this disease through its occurrence in the Royal family of Queen Victoria and its links to the Tsar empire and to the Spanish Royal family. However, documentation of this disease stems as long back as the 3rd century, described by the rabbins in the Talmud. Hemophilia A is inherited as an X-linked recessive disease expressed in males and with females being asymptomatic carriers.
This disease was shown to be connected with deficiency of a specific plasma protein in 1937, a protein which we denote as factor VIII but which was earlier called antihemophilic factor or antihemophilic globulin.
Hemophilia B is another bleeding disorder, caused by deficiency of factor IX, which was earlier denoted Christmas Factor.
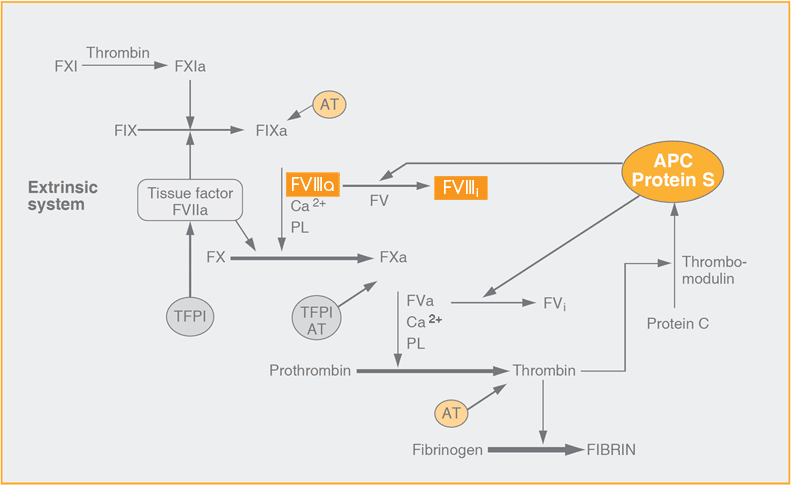
Coagulation is triggered by factor VII complexed with tissue factor from exposed subendothelium in the damaged vessel wall.
Factor VIII serves as a cofactor to the enzyme factor IXa in its activation of the zymogen factor X, other accessory components being calcium ions and procoagulant phospholipids expressed on the surface of activated platelets. Factor VIII must be activated before it can support factor IXa as an effective cofactor. In vitro such activation can be accomplished by thrombin and factor Xa but in vivo it may well be that thrombin is the only activator of physiological relevance to generate factor VIIIa. Under in vivo conditions, the rate increase of factor IXa activity due to factor VIIIa is several thousand-fold.
A similar rate increase of factor Xa activity is found for factor Va. It is no wonder, then, that the down-regulation of blood coagulation through specific inactivation of factor VIIIa and factor Va by activated protein C is a most efficient way of preventing further thrombin generation.
During the last decades new information has been gained on risk factors for venous and arterial thrombosis and the prevalence of genetic disorders among specific plasma components resulting in an increased risk. Examples on this are deficiencies of antithrombin, protein C and protein S and point mutations in factor V and prothrombin.
Factor VIII is a labile plasma component and determination of factor VIII activity must be made in a strictly controlled way to ensure accurate results. Blood sampling should be done with a clean venipuncture in order to avoid activation of blood coagulation and thereby the risk of inadvertent activation of factor VIII. Traditionally, clotting assays are used for assigning factor VIII activities, but chromogenic assays are increasing in use, especially in assigning potencies of factor VIII concentrates. The prime reasons for the gain in popularity is that chromogenic assays can be designed to provide robust assay conditions, offering a high resolution and without any negative interference from preactivated factor VIII molecules. These benefits have resulted in the selection of chromogenic methodology as the European Pharmacopoeia Reference method for factor VIII concentrates.
Biochemistry
The factor VIII gene and synthesis of the factor VIII protein
The factor VIII gene is located on the X chromosome and comprises no less than 26 exons separated by 25 non-coding introns and the complete gene constitutes about 0.1% of the chromosome.
The uncertainty about the site of synthesis and the low abundance of factor VIII, about 0.2 mg/L, made the cloning of the factor VIII gene a most challenging task. At the same time, it could be easily understood that the interest was considerable due to the low amount of protein needed to manage a proper hemostasis in hemophilia A patients. Indeed, and quite fascinating at the time, two groups simultaneously announced their successful cloning and expression of the factor VIII gene at the XVIth World Federation of Hemophilia Congress in Rio de Janeiro 1984, accompanied by publications in the same issue of Nature that year.
The site of synthesis of factor VIII has remained unclear because factor VIII mRNA has also been detected in many cells and tissues but it is most likely expressed in the hepatocytes. Independent support for the involvement of the hepatocytes is the beneficial effect of liver transplantation in severe hemophilia A patients.
Protein structure
The mature protein contains 2332 amino acids preceded by a 19-residue hydrophobic signal peptide of importance for the secretion.
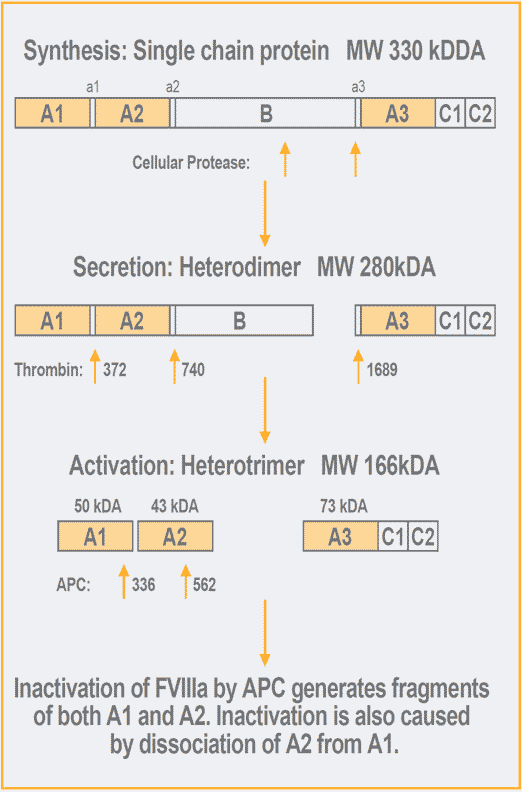
Factor VIII is synthesized as a single polypeptide chain with a molecular weight of about 330 kDa15 and it is thus an unusually large protein.
Factor VIII contains three different domains: an A-domain which is repeated three times, a central B-domain and a twice repeated C-domain. There are also small acidic peptide regions, one of which joins the A1 and A2 domains and another joining the B-domain with the amino terminal of the A3 domain. The overall structure has a high homology with factor V, which also is of a similar size. The amino acid sequence homology is, however, limited for the B-domains of the two proteins. In common, though, is that the B-domains are heavily glycosylated through asparagine linked carbohydrates.
Each of the A-domains share about 30% sequence homology with an A-domain structure also found in ceruloplasmin, a copper binding protein, and the C-domains are homologous to proteins which bind negatively charged phospholipids, the latter suggesting that these domains are important for the interaction with procoagulant phospholipids.
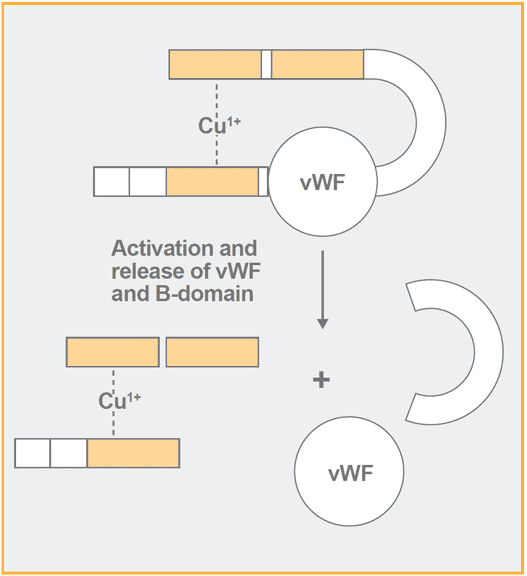
The amino-terminal signal peptide is removed upon translocation of factor VIII into the endoplasmic reticulum and the native factor VIII molecule is then cleaved in the B-domain in connection with its secretion. This results in the release of a heterodimer comprised of a 200 kDa heavy chain and an 80 kDa light chain, the association of which is metal-ion dependent.
During the secretion factor VIII associates with von Willebrand factor.
The homology of the A-domains with ceruloplasmin raised the question whether factor VIII may also be a copper-containing protein. This has been shown to be true for factor V and indeed it was more recently found to be true also for factor VIII and both contain 1 mole copper per mole of protein.
Data on Factor VIII
| Name: | Factor VIII |
| Synonyms: | Antihemophilic globulin |
| Gene location: | X-chromosome |
| Plasma concentration: | 0.2 µg/ml |
| Molecular weight: | Synthesized as 330 kDa single chain, secreted as two-chain 280 kDa molecule |
| Primary structure: | 2332 amino acids |
| Carbohydrate: | 5 % |
| Metal ion content: | One mole Cu1+ per mole protein |
| Half-life: | 12 hours, reduced severalfold in the absence of vWF |
| Function: | Cofactor to factor IXa in the activation of factor X |
| Importance: | Severe deficiency causes the bleeding disease hemophilia A Elevated activity is a risk factor for thrombosis |
Factor VIII also contains stabilizing disulphide bonds in the A and C domains as well as several sulfated tyrosines, claimed to be essential for maximal expression of factor VIII activity.
Interaction with von Willebrand factor
Since severe deficiency of vWF activity is associated with bleeding symptoms and low factor VIII activity, it was originally believed that factor VIII and vWF activities could be attributed to the same protein. Supporting this thought was the fact that early preparations of low purity factor VIII concentrates corrected the bleeding time. Furthermore, when purifying factor VIII from plasma, vWF was copurified and constituted more than 95% of the total protein content.
Negating both functions attributed one protein, von Willebrand’s disease is inherited as an autosomal disorder, whereas hemophilia A is an X-linked disease. Finally, in the 1970’s, convincing data was published detailing the non covalent association of factor VIII and vWF. This work allowed for a consensus on the identity of two proteins and roles.
Indeed, factor VIII is a trace protein in plasma with a concentration of only about 0.2 mg/L.
vWF serves an important role in targeting and concentrating factor VIII to the injured vascular wall via exposed subendothelium. When vWF is bound to factor VIII, it prevents binding of factor VIII to phospholipid surfaces including activated platelets and thereby factor VIII cannot be a part of the tenase complex with factor IXa, phospholipids and calcium ions. vWF also protects factor VIII from inactivation by activated protein C (APC) and from activation by factor Xa, but not by thrombin. The reason probably being that both factor Xa and APC require a phospholipid surface in their action on factor VIII whereas thrombin does not.
Activation of factor VIII
Factor VIII is activated by thrombin through specific proteolytic cleavages in both the heavy and the light chains. In the heavy chain one cleavage occurs at the amino acid Arg372 to generate separate A1 and A2 domains and another at Arg740 at the junction between the A2 domain and the B-domain resulting in the release of the B-domain.
In the light chain there is a cleavage at Arg1689 in the acidic region at the amino terminal of the A3-domain whereby a new NH2-terminus is created. Through this cleavage, vWF dissociates from factor VIII and the phospholipid binding region in the C-domains is exposed, which is mandatory for the cofactor activity of activated factor VIII (factor VIIIa). Interestingly, factor VIII contains two sulfated tyrosines at the positions 1664 and 1680 in the light chain, which are of importance for the binding of vWF.
Mutagenesis to obtain a phenylalanine at 1680 resulted in a loss of a high affinity vWF binding site31 and indeed the corresponding mutation in a patient resulted in moderate hemophilia.
The cleavages at 372 and 1689 are necessary for full activation of factor VIII. The 200 + 80 heterodimer is thus converted to a heterotrimer consisting of A1, A2 and A3-C1-C2. As mentioned above, the metal ion bridge is probably between the A1 and the A3-C1-C2 domains. Factor VIIIa has a molecular weight of about 165 kDa, derived from the A1 and A2 domains with molecular weights of 50 kDa and 43 kDa, respectively, and the 72 kDa light chain.
Factor VIII is also activated by factor Xa with cleavages occurring at the thrombin cleavage sites at 372 and 1689.
This suggests, but does not necessarily mean, that factor Xa is important for the physiological activation of factor VIII; it may be that the main role of the initial traces of factor Xa generated through the extrinsic pathway is to generate minute amounts of thrombin which then activates factor VIII.
The crucial importance of cleavages at 372 and 1689 for activation of factor VIII is also illustrated by the finding of severe hemophilia A patients with missense mutations at either of these residues. As might be expected, these patients had no measurable factor VIII activity but normal levels of factor VIII antigen.
Factor VIIIa is inherently unstable. Initially it was thought that the loss of activity was exclusively related to additional proteolysis by thrombin or factor Xa but it has later been shown to be much due to subunit dissociation. Thus, the loss of factor VIIIa procoagulant activity coincides with the dissociation of the A2 subunit from the factor VIIIa heterotrimer and the activity can also be restored by adding the A2 subunit to A2-deleted factor VIII. Addition of protease inhibitors does not prevent the loss of cofactor activity but addition of factor IXa and/or phospholipids improves the stability of human factor VIIIa. The latter finding may well be relevant in vivo with factor VIIIa bound to the membrane of activated platelets which have a large number, about 400 per platelet, of high affinity binding sites for factor VIII.
Cofactor activity of factor VIIIa in the tenase complex
Factor VIIIa serves as a most potent procoagulant cofactor in the tenase complex where factor X is activated by factor IXa in the presence of calcium ions and phospholipids. This is in analogy with the role of factor Va in the prothrombinase complex, comprising also factor Xa, calcium ions and phospholipids.
In vivo these processes take place primarily on the surface of activated and aggregated platelets which accumulate at the site of a vascular damage.
In contrast to resting platelets, activated platelets expose procoagulant phospholipids, such as negatively charged phosphatidylserine. Upon platelet activation, binding sites for both factors IXa, VIIIa, X /Xa, Va and prothrombin are exposed, which brings about a high local concentration of the constituents and an efficient generation of thrombin.
Furthermore, the cleavage in the factor VIII light chain and the release of vWF upon activation results in a considerably higher affinity for factor VIIIa to activated platelets as compared to unactivated factor VIII. In addition, factor X has been shown to increase the affinity of factor IXa for factor VIIIa approximately 10-fold. Both these features contribute to an optimal assembly of the components. Detailed studies have been performed on the interactions between factor VIII and factor IXa and it has been shown that the A3 domain in factor VIII interacts with the light chain of factor IXa and the A2 domain with the factor IXa protease domain.
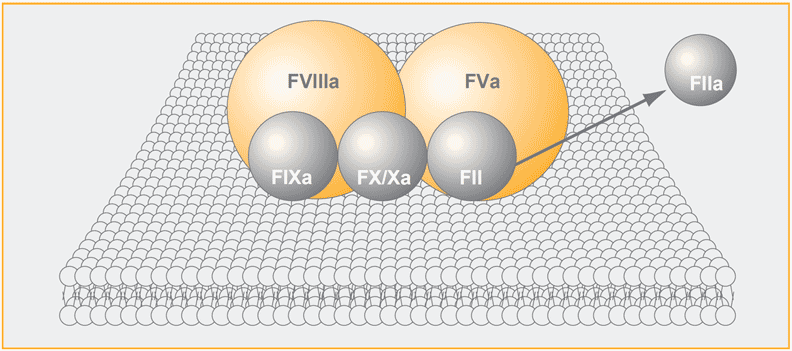
The tenase complex (factor IXa, VIIIa, X) and the prothrombinase complex (factor Xa, Va, prothrombin) assemble in close conjunction on the membrane surface of activated platelets in the presence of calcium ions. Factor Xa and thrombin are generated by the tenase and prothrombinase complex, respectively. The binding of factors IXa, X/Xa and prothrombin to the platelets is partly directed through Gla-domains in these proteins. Thrombin contains no Gla-domains and is released from the membrane whereafter it will cleave fibrinogen and other substrates.
The overall increase in the catalytic efficiency of factor IXa in the tenase complex as compared to factor IXa by its own, at physiological concentrations of reactants, is > 105 with factor VIIIa being responsible for a several thousand-fold increase of the reaction rate and where the highly increased affinity of factor X for factor IXa in the presence of procoagulant phospholipids is another major reason.
The effect of factor VIIIa is very similar to the effect of factor Va in the prothrombinase complex. As mentioned above, data have also been published which suggest that copper increases the cofactor activity of factor VIIIa.
| Factor X activator | Km factor X, µmol/L | Vmax, FXa x min-1 x IXa-1 |
|---|---|---|
| FIXa, Ca2+ | 181 | 0.01 |
| FIXa, Ca2+, PL | 0.36 | 0.025 |
| FIXa, Ca2+, PL, FVIIIa | 0.29 | 500 |
The physiological triggering of coagulation is most probably caused by the extrinsic pathway whereby factor VII is efficiently activated after complexing with exposed tissue factor (TF) whereupon factor IX and factor X are activated by the factor VIIa/TF complex. It is a fact that hemophiliacs bleed severely and thus the clinical evidence clearly shows that activation of factor X by the tenase complex is of great importance in vivo. Thus, the factor VIIa/TF complex is downregulated by TFPI after binding factor Xa and hence continued direct activation of factor X is inhibited. Furthermore, the activation of factor X by the factor IXa-factor VIIIa pathway is about 50-fold more efficient than the direct FX activation by factor VIIa/TF.
The contribution by the factor IXa pathway is also efficiently promoted by factor XIa, which activates factor IX.
The findings that thrombin as well as meizothrombin activate factor XI and that such activation also can occur on the platelet surface indicate that this event probably is of importance in vivo. The activation of factor XI by thrombin may thus result in an increased generation of factor IXa, supporting further the important role of factor VIII in obtaining a sufficient generation of thrombin. Indeed, this additional positive feed-back role of thrombin serves as a further beautiful illustration of how thrombin regulates its own activity and fate.
Proteolytic inactivation of factor VIIIa by APC
A most efficient down-regulation of coagulation is provided by APC, which inactivates factor Va and factor VIIIa through specific proteolytic cleavages. Within factor VIIIa, these cleavages occur at Arg336 in the A1 subunit and Arg562 in the A2 subunit. It seems that in the human system, the cleavage at 336 is the preferred initial cleavage site with concomitant loss of cofactor activity.
There are fascinating interactions between the components of the tenase system and of the protein C anticoagulant system which affect the rate of inactivation of factor VIIIa. Thus, factor IXa protects the cleavage by APC at Arg562 and factor X protects the 336 cleavage site. However, protein S abrogates the protecting effect of factor X and, in the absence of factor IXa, also stimulates the rate of cleavage at 562 considerably. Corresponding regulatory roles for factor Xa and protein S have also been demonstrated for the APC-dependent inactivation of factor V.
The relative importance of degradation of activated factors V and VIII by APC on the down-regulation of coagulation is not clear. Some data indicate that inactivation of factor Va is the crucial effect of APC and it has also been shown in mutagenesis studies that both APC cleavage sites had to be blocked before a significant impairment of the rate of thrombin generation was registered in an APTT-based method and that indeed the major inactivation of factor VIIIa may be caused by dissociation of the A2 subunit.
Clinical Aspects
Hemophilia A background and classification
Hemophilia A is a serious bleeding disorder which is caused by a deficiency or complete absence of factor VIII activity which affects about 1 in 5000 males and its prevalence show no ethnic differences.
It is inherited as an X-linked disorder but in many cases there is no family history simply due to the fact that about 30% of patients have a recent, spontaneous mutation. The obvious bleeding symptoms mean that this disorder, initially named haemorraphilia (love of bleeding) can be traced back to very early observations and in 1835 the disease was described as a protein disorder. Within only a few years later the first blood transfusion was administered to a patient for treatment of his bleeding.
Since then, tremendous progress has been made in the treatment of hemophilia patients who typically now receive highly purified plasma factor VIII or recombinant factor VIII concentrates. This change in treatment provides a minimal risk of transmittance of viral infections previously seen with blood transfusions.
| Factor VIII activity | Clinical Hemophilia Classification |
| < 0.01 IU/mL (< 1% of normal) | severe |
| 0.01 – 0.05 IU/mL | moderate |
| > 0.05 – < 0.40 IU/mL | mild |
| Classification of hemophilia A The bleeding disorder hemophilia A is classified into three categories depending on the level of factor VIII activity. | |
Depending on the factor VIII activity, which is related to the bleeding severity, hemophilia A is classified according to the table above. However, it is not a straightforward relationship. There are patients with a factor VIII level < 1% who have very little or no spontaneous bleedings, while spontaneous and clinically severe bleeding occur in some patients with 1-5% factor VIII activity. Since patients with von Willebrand’s disease also have a decreased factor VIII activity, a correct diagnosis of hemophilia A requires determination of vWF antigen or ristocetin cofactor activity. vWF antigen and activity levels are normal in hemophilia patients. Furthermore, the family history also provides important information since von Willebrand’s disease is inherited as an autosomal trait. A rare variant, Type 2N, of von Willebrand´s disease in which vWF does not bind to factor VIII, may be misdiagnosed for hemophilia A. To distinguish Type 2N of VWD, vWF-factor VIII binding studies must be performed.
Severe hemophilia A patients typically meet with bleedings in joints and muscles and sustained and dangerous bleedings after trauma and surgery and these patients may, unless treated, develop permanent disability. In moderate or mild hemophilia A bleeding episodes are more rare and occur usually in connection with trauma or surgery.
Clinical management of Hemophilia
History
The only available treatments for hemophilia A patients for many decades were whole blood or plasma but this was often not sufficient to rapidly achieve proper hemostasis. This situation changed dramatically after it was discovered in 1956 that factor VIII coprecipitated with fibrinogen, in the so called Cohn fractionation system. The same year, a crude factor VIII-vWF preparation was administered to a woman with severe von Willebrand´s disease. This preparation was also rapidly introduced for treatment of hemophilia A patients.
After this pioneering work a further important step forward was the discovery that factor VIII is recovered in plasma cryoprecipitate, which allowed an effective replacement therapy. Still, though, the specific activity was low, about 0.5 IU/mg, the major proteins being fibrinogen and fibronectin.
More pure factor VIII preparations, so called intermediate purity concentrates, were developed over the years using different types of chromatography, which resulted in specific activities of up to 10 IU/mg.
The next step forward in purification was the introduction in the late 70s. Immunoaffinity purification of factor VIII was produced using matrix-bound antibodies against vWF and elution of factor VIII with calcium ions.
Within a decade immunoaffinity techniques were used for large-scale purification of factor VIII whereby factor VIII-specific antibodies also were utilized and specific activities close to the theoretical maximum of about 5000 IU/mg could be obtained. However, due to the labile nature of factor VIII, albumin is usually added as a stabilizer, which results in specific activities of about 15 IU/mg.
The dosages of factor VIII concentrate used depend upon the specific bleeding event, but normally the factor VIII level must exceed 0.3 IU/mL in plasma in order to achieve normal hemostasis; in acute serious trauma about 1 IU/mL may be needed.
This requires the infusion of about 15-40 IU/kg bodyweight. Repeated infusions are necessary to maintain sufficient factor VIII levels since the half-life of factor VIII in vivo is only about 12 h. The advent of Extended Half Life (EHL) products has helped, by increasing the time required between dosing.
Much lower doses are used for prophylaxis, whereby the target factor VIII levels are above 0.03 IU/mL.
The availability and introduction of factor VIII concentrates for treatment of acute bleedings and for prophylaxis has had a dramatic improvement of the life expectancy of severe hemophilia A patients. In parallel, the development and progression of joint disease has decreased significantly.
The use of blood products is connected with a risk of viral transmission such as hepatitis B and C, and many hemophiliacs developed chronic liver disease but the great benefits of the therapy was considered to justify the treatment.
The picture changed dramatically from 1982 when the first hemophilia A patient was identified with AIDS as a result of infusion with factor VIII concentrate. A cruel decade followed with thousands of hemophilia patients killed by AIDS due to HIV virus contamination in factor VIII concentrates. This was a tragedy not only to the patients and their families but certainly also to all clinicians and other medical staff who showed a great engagement and commitment to good hemophilia care.
Development of concentrates with maximal safety against viral transmission was made by two routes, which luckily has resulted in no new HIV infections of hemophilia patients.
One route was through introduction of extensive heat treatment or use of solvent-detergent treatment of plasma derived factor VIII concentrates, the other through the development of recombinant factor VIII concentrates. From the cloning of the factor VIII gene in 1984, ten years passed until recombinant factor VIII concentrates were registered for clinical use, preceded by publications on their safe and successful applications.
Later, a recombinant truncated form of factor VIII (ReFacto®, Pfizer, lacking most of the B-domain, was developed and characterized and shown to be equally clinically effective. This preparation has no addition of human albumin as a stabilizer, thereby showing a possibly even lower risk of viral transmission. In mild and moderate hemophilia A patients, sufficiently high factor VIII activity levels can be reached in most patients by administering desmopressin, an analogue to the diuretic hormone vasopressin.
This agent has not only the benefit of having no risk of viral transmission but it can also be provided to the patient intranasally.
Extended Half Life Products
More recently, modified recombinant FVIII replacement products have been introduced, with the benefit of increasing the time needed between doses. The modified structure and function of these EHL’s have resulted in assay discrepancies, with different aPTT and instrument combinations giving a variety of results. The coagulation laboratory must now be more aware of these different assay pros and cons. There is a large and growing body of publications on this topic as time goes on.
Emicizumab
Emicizumab (Hemlibra®) is a recombinant bispecific monoclonal antibody that restores the function of the missing FVIII. It acts by physically bridging FIXa and FX. Essentially, it mimics native FVIII function. Emicizumab interferes with some coagulation assays, like aPTT, and care must be taken to choose appropriate assays. Chromogenic FVIII assays containing bovine proteins (Factor Reagent) that don’t interact with, thus, detect emicizumab. This enables the assay to specifically measure endogenous or infused FVIII activity, without interference of the activity/level of emicizumab itself.
Other avenues
While replacement therapy and/or emicizumab are the current treatment options, exciting innovations are under development to improve treatment and outcomes. These include recombinant clotting factors, bioengineered factors, and drugs targeting other pathways to increase coagulation, such as Tissue Factor Pathway Inhibitor (TFPI). Gene therapy is the latest treatment angle to be approached. Gene therapy strategies have an end goal of increasing long-term expression of Factor VIII, ultimately reducing or even eliminate the need for other treatments.
Factor VIII inhibitors
Unfortunately, a portion of hemophilia A patients develop antibodies against factor VIII, denoted as “factor VIII inhibitors”. This occurs in 5-15% of patients with mild or moderate severity and about 25% of patients with severe hemophilia A after being treated with factor VIII concentrates. The titer of factor VIII inhibitor antibodies is reported in Bethesda units, defined by a specified test system (see below). This titer can vary greatly between patients ranging from 0.5 -15.000 units. Patients are classified as high or low responders and it has recently been decided to use the term high responder for a patient who at any time presents with an antibody titer above 5 Bethesda units whereas patients who persistently have below 5 Bethesda units, despite repeated treatments with factor VIII concentrates, are denoted as low responders.
Does the type of mutation affect the risk of developing factor VIII inhibitors? Yes, patients with large deletions and nonsense mutations or gene inversions develop inhibitors to a larger extent than those with frameshift or missense mutations. It also seems that the greatest risk of raising inhibitors is during the initial treatment.
Does the type of factor VIII concentrate affect the development of inhibitors? Generally, no. Although, it has been reported that modification of a concentrate during manufacturing resulted in antibody development in a patient. On the other hand, the type of concentrate to choose for treatment may play a role. Thus, if a patient has developed antibodies against the light chain of factor VIII, it may be preferable to administer a concentrate which is rich in vWF, since factor VIII then appears to be more slowly neutralized and hence more efficient. Since, high responding patients may meet with life-threatening conditions, other treatment regimens have been developed. These alternatives included high dosage porcine factor VIII concentrate and immunosuppression therapy. This also seems to be one way of inducing immune tolerance in high responding patients, a desired but difficult goal and which is more often obtained in low-responding patients. Sometimes by “merely” infusing high doses of factor VIII this goal can also be reached with low-responders. A third treatment regime with rapid achievement of normal hemostasis in many instances is infusion of recombinant factor VIIa. This will probably gain increased use in the future.
Further, Emicizumab (Hemlibra®), described above, is indicated for hemophilia patients with and without FVIII inhibitors.
When measuring FVIII inhibitors in patients on Emicizumab, a chromogenic FVIII Bethesda assay can be used, as long as it is made with bovine coagulation proteins. Clotting-based Bethesda assays should not be used, as they can give false negative inhibitor titers. Since Emicizumab only reacts in a FVIII chromogenic substrate assay if human factors are used, measurement of endogenous FVIII and FVIII inhibitors is possible with chromogenic assays that employ non-human factor reagents, as this allows one to measure residual FVIII activity. The inhibitor concentration is determined using an equation that relates the log % residual activity to inhibitor titer.
The future – gene therapy?
Cost is a prime issue in the treatment of hemophilia and it is a main obstacle in providing proper treatment worldwide. Thus, the prospects of bringing efficient, modern treatment into global use are still challenging.
Since only minute amounts of factor VIII need to be present in plasma for proper hemostasis, great efforts are being put in gene therapy research. The real challenge, apart from important safety issues, is to achieve a sustained production of factor VIII at a low level. The first attempts were made in the early 90s and now a number of different approaches are being explored, including retroviral, adenoviral and non-viral gene deliveries along with utilizing different target cells.
The goal of gene therapy is to provide long-term expression of Factor VIII, to reduce or even eliminate the need for other treatments like factor replacement or emicizumab. With a single IV infusion, a modified adeno-associated virus (AAV) that targets liver cells acts as the vector to deliver a copy of functional FVIII gene, enabling the body to make Factor VIII. As is, AAV capsid immune response and liver toxicity issues must be addressed. Clinical trials are ongoing.
Assay discrepancies between one stage and chromogenic substrate assays have been demonstrated in gene therapy trials. The reasons for these discrepancies and solutions to them are the focus of numerous current studies.
Elevated factor VIII activity as a risk factor for thrombosis
It has long been known that arterial and venous thrombosis are multifactorial diseases. Thus, combined abnormalities of factor V:Q506 (factor V Leiden) and inherited deficiency of either of antithrombin, protein C or protein S, results in a significantly higher incidence of venous thrombosis. Studies now also support that elevated factor VIII levels are an important risk factor as well.
In 1980 a prospective study indicated factor VIII to be a risk factor for arterial disease and other studies also suggested association of elevated factor VIII with both cardiac and cerebral vascular disease and increased morbidity or earlier fatal outcome. This was later supported in mouse studies with controlled mild carotid artery injury and received infusion of factor VIII, which suggested a direct thrombogenic role for factor VIII.
In 1995 elevated factor VIII activity was shown to be an independent, highly prevalent risk factor with an odds ratio of up to 6 and it is recommended to be included in the laboratory screening test panel on analysis of plasma from thrombotic patients.
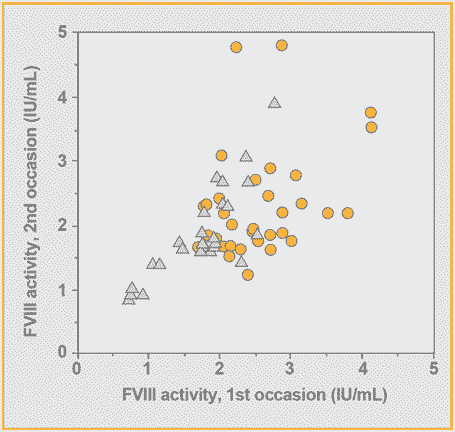
A comparison of results from a second determination of factor VIII activity made between 3 months and 4 years after the first determination in patients with venous thromboembolism.
In more than 20% of the patients, factor VIII activities were >1.5 IU/mL and occasionally levels as high as 4-5 IU/mL were found. There was also a close correlation to factor VIII antigen, demonstrating that there was an increased synthesis of the factor VIII protein and thus the rise in activity is not due to generation and circulation of activated factor VIII. However, factor VIII is an acute phase reactant and there were, quite understandable, early doubts as to whether elevated factor VIII levels have any causal role but perhaps rather were an effect of the disease.
Thus, elevated levels are associated with conditions such as trauma, infection, and exercise and, in common to many other coagulation factors, factor VIII activity is also increased during pregnancy.
It was shown, however, that elevated factor VIII activities were not linked to any acute response and that they were indeed persistent with similarly high levels demonstrated upon repeated analysis after 3 months to 4 years.
In conclusion, there is now a substantial amount of data which points to a causal role for elevated factor VIII activity and at least venous thrombosis and it should be expected that analysis of factor VIII activity will be increasingly introduced in routine laboratory investigations of thrombotic patients.
Determination of factor VIII activity
The one-stage clotting method
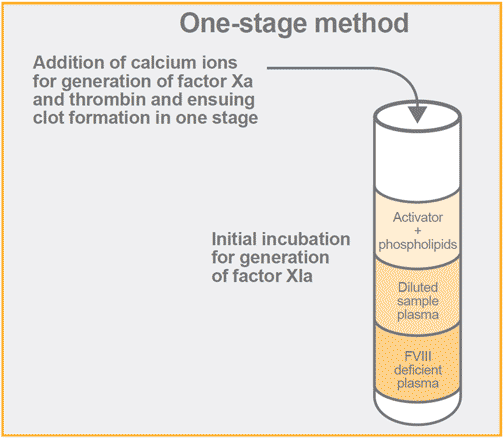
The most widely used method for factor VIII activity determination in plasma is the one-stage clotting method. This method is based upon the activated partial thromboplastin time (APTT) used with congenital severe hemophilia A plasma or artificially prepared factor VIII deficiency plasma as a substrate.
This method was developed in the early 50s and is based on the ability of a plasma sample to shorten the prolonged APTT of factor VIII deficient plasma. In principle, the effect is related to the amount of factor VIII activity in the sample. In the assay system, phospholipids are added to citrated plasma along with a negatively charged surface activator, e.g. kaolin, which thereby activates the contact factors and leads to the generation of factor XIa. Upon addition of calcium ions, factor IX is activated by factor XIa and factor Xa is then generated by the tenase complex (intrinsic system), in which factor VIII serves as a cofactor. The time for fibrin clot formation, due to cleavage of fibrinogen by generated thrombin, is recorded. A standard curve is constructed from assaying different dilutions of a normal plasma with a known factor VIII activity. Results are most commonly express in a double logarithmic plot with log factor VIII activity vs log clotting time. The factor VIII activities of assayed plasma samples are then derived from the standard curve.
The one-stage method has the advantage of being rapid and easy to perform. At the same time, it must be realized that the assay is deceptively simple since it indeed comprises a complex biochemical system with a short total reaction time. The shortcomings include an increased sensitivity to preactivated clotting factors, which will result in overestimation of factor VIII activity, and interference by Lupus anticoagulants. It is also clear from several surveys that there is an appreciable variability in results, with interlaboratory coefficients of variation sometimes reaching 45%. Some of this variability could be explained by improper standardization and less satisfactory instrument performance.
With the introduction of international standards of plasma and factor VIII concentrate and with the increasing use of automated coagulation instruments, the situation has improved but there is still a considerable variability which probably can be explained using different sources of phospholipid, contact activator and factor VIII deficiency plasma.
It is also possible that the increasing use of prophylaxis for severe hemophilia A patients will limit the availability of congenital factor VIII deficiency plasma and therefore the quality of artificially prepared factor VIII deficiency plasma may be of increasing importance.
The two-stage clotting method
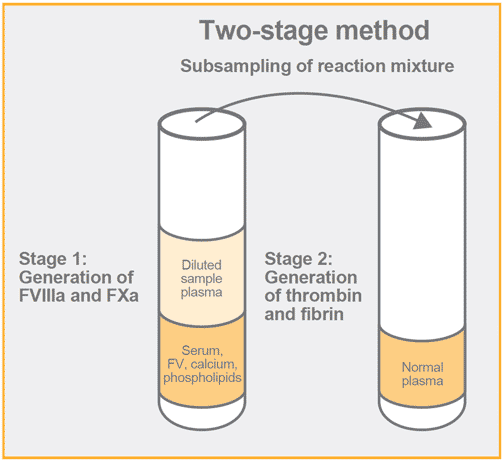
This method was also developed in the 50s and it is based upon a first step whereby activated factors V and X are generated in an amount, which is related to the sample factor VIII activity. In a second step, prothrombin and fibrinogen are added, usually in the form of normal plasma, and the clotting time is recorded.
This method makes no use of factor VIII deficiency plasma. It is claimed to show a better precision in general and to be more sensitive than the one-stage method. It is also less prone to interference than the one-stage method and its design makes it insensitive to preactivation of factor VIII.
These properties resulted in the two-stage method being selected as the reference method for determination of the factor VIII potency of factor VIII concentrates for several decades. However, it is more cumbersome to perform, and it appears that expert laboratories are best suited to explore the full potential of the method; hence it is not commonly used today.
Additional factor VIII clotting methods
Other approaches than the one-and two stage clotting methods have also been utilized, such as different thrombin generation tests. These vary from straightforward recalcification of plasma to more elaborate methods in which partially purified factors were utilized. With the use of lyophilized mixtures of well-defined reagents, the thrombin generation test was, however, made much more convenient and seemingly suitable for routine use. None of these methods, however, gained widespread use.
Chromogenic methods
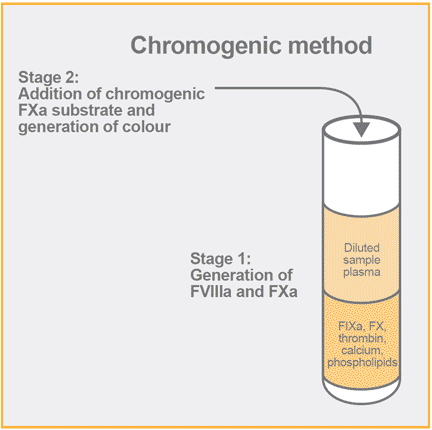
With the invention of chromogenic substrates, a new avenue was opened for designing methods for hemostatic factors and their general suitability for determination of enzymes, proenzymes, inhibitors and cofactors was shown as well as their applicability to automated instruments.
The availability of the highly selective chromogenic substrate S-2222 (Benzoyl-Ile-Glu-Gly-Arg-pNA) for factor Xa triggered the development of chromogenic methods for factor VIII activity and a specific method with a high resolution and with stabilized reagents was published in 1983.The assay principle is like the two-stage assay in that is starts with an incubation step for generation of factor Xa. Furthermore, there is no need of factor VIII deficiency plasma, rather the assay also utilizes purified bovine factors IXa and X. In contrast to the two-stage assay, instead of using a fibrin clotting time as the end-point, the amount of factor Xa is determined from the hydrolysis of the FXa substrate. In this case, the substrate is known as S-2222. Future kit improvements made use of another FXa substrate S-2765.
In the chromogenic assay, the amount of generated factor Xa is directly proportional to the factor VIII activity producing a high-resolution test. This feature makes the method suitable for both measurement of very low levels, such as in the classification of hemophilia A patients, as well as of very high levels by proper adjustment of sample dilution and incubation time. Highly elevated levels of factor VIII (< 4 IU/ml) can be determined with maintenance of a high accuracy by making an initial sample predilution. The chromogenic method correlates closely with the one-stage clotting method, including recovery studies after administration of factor VIII concentrate to hemophilia A patients, and like the two-stage method, it was demonstrated to be insensitive to preactivation by thrombin. The high dilution used in the chromogenic method as compared to clotting methods also means that interferences can be minimized. One illustration of this is the lack of influence of Lupus anticoagulants, whereby the added procoagulant phospholipid override the inhibitory effect of anti-protein-phospholipid antibodies. In addition to clinical applications, the chromogenic factor VIII assay has proven to add utility and value to other applications in manufacturing and the research fields. It is shown to be suitable for analysis of factor VIII concentrates and for screening of blood donors. This method can be adapted by performing the analyses in microplates, thereby allowing a high throughput, precision, and a cost-competitive analysis as compared to the one-stage method. Moreover, the high sensitivity of the chromogenic method is suitable for detection of recombinant expression of factor VIII in cell culture conditions and has proven valuable in research studies on factor VIII activity using purified factor VIII and of separate or combined subunits. Clinical research has utilized the chromogenic factor VIII assay for studies on the effect of factor VIII activity after intranasal administration of DDAVP.
Other variants of the chromogenic method have since been developed and commercialized utilizing human factors IXa and X or by adding thrombin to obtain an immediate activation of factor VIII with shorter assay incubation times. This has allowed the chromogenic method to be conveniently applied on automated instruments.
Diagnosis and classification of hemophilia A
Both one-stage, two-stage and chromogenic methods are today used for diagnosis and classification of hemophilia A patients. At the present time, most of the tests performed use the one-stage method.
The fact is that patients with the same factor VIII activity as measured with one method may show quite different clinical symptoms.
Case reports demonstrate a lack of clinical accuracy for different tests under different circumstances. While each test might have advantages and disadvantages, clinicians should utilize all options available for the most complete picture.
More recently, several studies have confirmed the discrepancy between one-step and chromogenic substrate methods, particular with the enhanced half-life products and gene therapy as previously discussed.
Thus, similar as for other hemostasis analytes, there seems to exist no method which can safely claim to reflect accurately the clinical picture for all hemophilia A patients. A humble attitude is therefore recommended which should encourage the clinician to use more than one method when the first line results are not in concordance with the clinical severity.
Determination of factor VIII inhibitor activity in plasma
As mentioned above, about 25% of severe hemophilia A patients develop factor VIII inhibitors.
The proper treatment of these patients requires correct determination of the inhibitor titer.
Initially, incubations were made of patient plasma with factor VIII concentrate (the New Oxford method). This original approach with the Oxford method was prob but the instability of factor VIII activity in concentrates and the lack of standardization, resulted in the development and acceptance of another method, called the Bethesda method. For the Bethesda method, patient plasma is incubated with normal plasma for a defined time followed by determination of the residual factor VIII activity. Usually clotting time determination is used as the end-point but the applicability of chromogenic methods have also been shown for both the New Oxford and Bethesda methods.
Despite the introduction of an international standard for plasma factor VIII activity, the interlaboratory variability of factor VIII inhibitor determinations had remained significant. The use of buffered normal plasma as FVIII source increased the stability of the factor VIII activity, which ultimately improved reliability and specificity of the test. The benefits of this approach, denoted the Nijmegen modification, were later demonstrated in a large study of close to 900 patient samples.
The Nijmegen-Bethesda method has been successfully used in a chromogenic application in connection with clinical studies related to hemophilia A patients.
Determination of factor VIII concentrate potency
Discrepancies between methods for determination of factor VIII potency in factor VIII concentrates was already a concern with low or intermediate purity concentrates, but this concern persisted for plasma concentrates with higher purity in spite of the introduction of international concentrate standards. The introduction of such standards in combination with harmonizing assay conditions has gradually resulted in more reliable assays. Important improvements include the predilution of the concentrate to 1 IU/mL in factor VIII deficiency plasma, as well as the incorporation of high quality bovine or human albumin in the buffer used for final dilutions. Subsequent multicenter studies confirmed the beneficial effect obtained by these modifications.
The two-stage clotting method had previously been the European Pharmacopoeia reference method for factor VIII potency determination of concentrates for several decades. Although the one-stage method is easier to perform, it was not selected as the reference method due to the interferences in this method from lipids or traces of heparin and its sensitivity to preactivation of factor VIII. Indeed, substantially higher potencies (up to 40%) were obtained with the one-stage method.
The chromogenic method has a principal similarity to the two-stage method and a high agreement with this method was also obtained in several labs in potency determinations of concentrates. This feature together with its higher precision resulted in the recommendation of the ISTH Subcommittee for factor VIII and factor IX to adopt the chromogenic method as the reference method and it was later also selected as the reference method by the European Pharmacopoeia.
When measuring Extended Half-Life Products and investigating Gene Therapy, care must be taken to consider these known assay discrepancies. Further discussion can be found in our inaugural Clot Club post, here.
Work remains until concordant results are obtained between clotting, especially one-stage, and chromogenic methods. In the meantime, it seems to be the consensus to dose hemophilia A patients with factor VIII concentrates that have potency assignments according to the chromogenic method for a safe and cost-effective treatment.
Summary
The last century has showed a most impressive increase in our knowledge of factor VIII including biochemistry, methodology and the treatment of hemophilia A patients. This fascinating protein has rightly attracted, and continues to attract, researchers and clinicians. The 20th century saw the launch of factor VIII concentrates, with high safeguarding against transmittal of infectious diseases, and the 21st century has seen new therapies and promises of gene therapy in the not too distant future.
It remains an important task to make modern treatments more accessible in the majority of countries to the benefit of the hemophilia A patients.
