Coagulation (clotting) is the process by which blood changes from a liquid to a gel, forming a clot. It potentially results in hemostasis, the cessation of blood loss from a damaged vessel, followed by repair. The mechanism of coagulation involves activation, adhesion, and aggregation of platelets along with deposition and maturation of fibrin. Disorders of coagulation can result in bleeding (hemorrhage or bruising) or obstructive clotting (thrombosis).
Coagulation Cascade
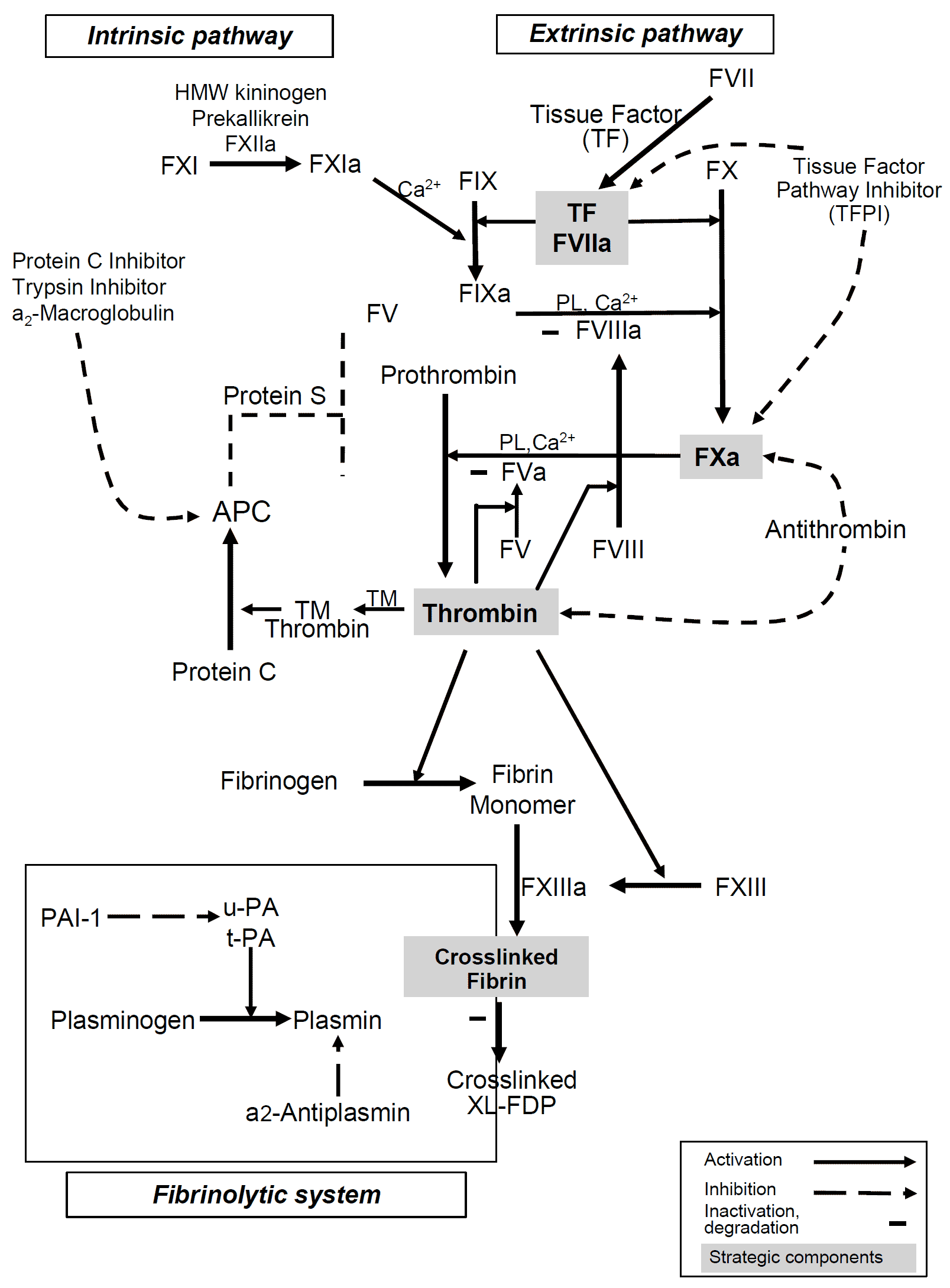
The coagulation cascade of secondary hemostasis has two initial pathways which lead to fibrin formation:
- contact activation pathway (intrinsic pathway)
- tissue factor pathway (extrinsic pathway)
The primary pathway for the initiation of blood coagulation is the tissue factor (extrinsic) pathway. Both the tissue factor and contact activation pathways both activate the “final common pathway” of factor X, thrombin and fibrin.
The pathways are a series of reactions, in which a zymogen (inactive enzyme precursor) of a serine protease and its glycoprotein co-factor are activated to become active components that then catalyze the next reaction in the cascade, ultimately resulting in cross-linked fibrin.
The coagulation factors are generally serine proteases (enzymes), which act by cleaving downstream proteins, with some exceptions. FVIII and FV are glycoproteins, and Factor XIII is a transglutaminase. The coagulation factors circulate as inactive zymogens.
Tissue Factor Pathway (Extrinsic Pathway)
The main role of the tissue factor pathway is to generate a “thrombin burst”, a process by which thrombin, the most important constituent of the coagulation cascade in terms of its feedback activation roles, is released very rapidly. FVIIa circulates in a higher amount than any other activated coagulation factor. The process includes the following steps:
- Following damage to the blood vessel, FVII leaves the circulation and comes into contact with tissue factor (TF) expressed on tissue-factor-bearing cells (stromal fibroblasts and leukocytes), forming an activated complex (TF-FVIIa).
- TF-FVIIa activates FIX and FX.
- FVII is itself activated by thrombin, FXIa, FXII and FXa.
- The activation of FX (to form FXa) by TF-FVIIa is almost immediately inhibited by tissue factor pathway inhibitor (TFPI).
- FXa and its co-factor FVa form the prothrombinase complex, which activates prothrombin to thrombin.
- Thrombin then activates other components of the coagulation cascade, including FV and FVIII (which forms a complex with FIX), and activates and releases FVIII from being bound to vWF.
- FVIIIa is the co-factor of FIXa, and together they form the “tenase” complex, which activates FX
Contact Activation Pathway (Intrinsic Pathway)
The contact activation pathway begins with formation of the primary complex on collagen by high-molecular-weight kininogen (HMWK), prekallikrein, and FXII (Hageman factor).
- Prekallikrein is converted to kallikrein and FXII becomes FXIIa.
- FXIIa converts FXI into FXIa.
- Factor XIa activates FIX, which with its co-factor FVIIIa form the tenase complex, which activates FX to FXa.
The minor role that the contact activation pathway has in initiating clot formation can be illustrated by the fact that subjects with severe deficiencies of FXII, HMWK, and prekallikrein do not have a bleeding disorder. Instead, contact activation system seems to be more involved in inflammation, and innate immunity.
Final Common Pathway
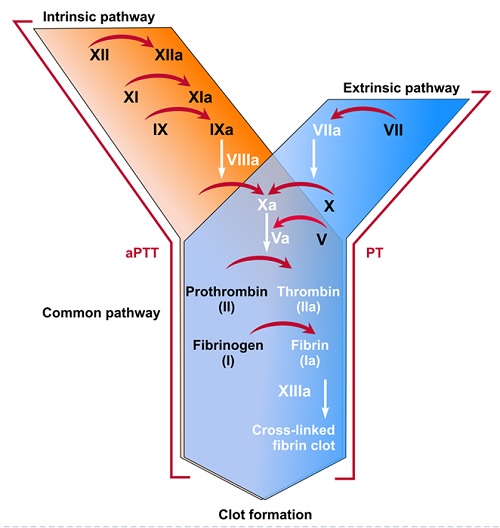
Following activation by the contact factor or tissue factor pathways, the coagulation cascade is maintained in a prothrombotic state by the continued activation of FVIII and FIX to form the tenase complex, until it is down-regulated by the anticoagulant pathways.
Coagulation Factors
Factor / name |
Alternate name |
Function |
Associated Genetic Disorders |
|---|---|---|---|
| Alpha 2-Antiplasmin | α2-antiplasmin, plasmin inhibitor | Inhibits plasmin | Antiplasmin deficiency |
| Antithrombin | Antithrombin III, AT, ATIII | Inhibits Factor IIa, Xa, and other proteases | Antithrombin III deficiency |
| Cancer Procoagulant | Pathological factor X activator | linked to thrombosis in cancer | |
| Factor I | FI, Fibrinogen | Blood clot formation (Fibrin) | Congenital afibrinogenemia, Familial renal amyloidosis |
| Factor II | FII, Prothrombin | Factor IIa activates I, X, VII, VIII, XI, XIII, protein C, platelets | Prothrombin G20210A, Thrombophilia |
| Factor III | FIII, Tissue Factor, Tissue Thromboplastin | Co-factor of VIIa (formerly known as Factor III) | |
| Factor IV | FIV, Calcium | Required for coagulation factors to bind to phospholipid (formerly known as factor IV) | |
| Factor V | FV, Proaccelerin, Labile factor | Cofactor of X with which it forms prothrombinase complex | Activated protein C resistance |
| Factor VI | FVI, Factor Va | Unassigned | |
| Factor VII | FVII, Stable Factor, Proconvertin | Activates Factor IX, X | Congenital proconvertin / Factor VII Deficiency |
| Factor VIII | FVIII, Antihemophilic Factor A, AHF | Cofactor of IX with which it forms tenase complex | Haemophilia A |
| Factor IX | FIX, Antihemophilic factor B, Christmas Factor | Activates Factor X, forms tenase complex with Factor VIII | Haemophilia B |
| Factor X | FX, Stuart-Prower Factor | Activates Factor II, forms prothrombinase complex with Factor V | Congenital Factor X deficiency |
| Factor XI | FXI, Plasma Thromboplastin Antecedent | Activates Factor IX | Haemophilia C |
| Factor XII | FXII, Hageman Factor | Activates Factor XI, VII and Prekallikrein | Hereditary angioedema type III |
| Factor XIII | FXIII, Fibrin-Stabilizing Factor | Crosslinks Fibrin | Congenital Factor XIIIa/b deficiency |
| Fibronectin | Mediates cell adhesion | Glomerulopathy with fibronectin deposits | |
| Heparin Cofactor II | HCII | Inhibits Factor IIa, cofactor for heparin and dermatan sulfate (“minor antithrombin”) | Heparin cofactor II deficiency |
| High Molecular Weight Kininogen | HMWK, HK, Fitzgerald Factor | Supports reciprocal activation of Factor XII, XI, and prekallikrein | Kininogen deficiency |
| Plasminogen | PLG | Converts to plasmin, lyses fibrin and other proteins | Plasminogen deficiency type I (ligneous conjunctivitis) |
| Plasminogen Activator Inhibitor-1 | PAI1, PAI-1 | Inactivates tPA & urokinase (endothelial PAI) | Plasminogen activator inhibitor-1 deficiency |
| Plasminogen Activator Inhibitor-2 | PAI2, PAI-2 | Inactivates tPA & urokinase (placental PAI) | |
| Prekallikrein | Fletcher Factor | Activates Factor XII and prekallikrein; cleaves HMWK | Prekallikrein/Fletcher Factor deficiency |
| Protein C | PC, autoprothrombin IIA, Factor XIV | Inactivates Factor Va and VIIIa | Protein C deficiency |
| Protein S | PS | Cofactor for activated protein C (APC, inactive when bound to C4b-binding protein) | Protein S deficiency |
| Protein Z | PZ, PROZ | Mediates thrombin adhesion to phospholipids and stimulates degradation of factor X by ZPI | Protein Z deficiency |
| Protein Z-related Protease Inhibitor | ZPI | Degrades Factors X (in presence of protein Z) and XI (independently) | |
| Tissue Plasminogen Activator | tPA | Activates plasminogen | Familial hyperfibrinolysis and thrombophilia |
| Urokinase | UK | Activates plasminogen | Quebec platelet disorder |
| von Willebrand Factor | vWF | Binds to Factor VIII, mediates platelet adhesion | von Willebrand disease |
Cofactors
- Calcium and phospholipid
- Vitamin K
Regulators
- Protein C
- Antithrombin
- Tissue factor pathway inhibitor (TFPI)
- Plasmin
- Prostacyclin (PGI2)
View the Diapharma special coagulation presentation