Proposed Mechanisms of Platelet Dynamics during COVID-19
Posted on: October 2, 2020

– Contributed by Abi Kasberg, PhD
The novel coronavirus worldwide pandemic of 2020 is infecting and claiming lives at a rapid rate. Infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) can lead to severe acute respiratory distress and multiple system organ failure in a disease called COVID-19. SARS-CoV-2 enters cells by endocytosis through binding of the viral spike protein to the cell membrane enzyme ACE2. It is predicted that the main entry point of SARS-CoV-2 into the body is through the respiratory system due to the expression of ACE2 on airway epithelium and lung tissue. Following initial respiratory damage, clinical reports suggest that in a subgroup of severe COVID-19 patients the immune system responds with high levels of cytokine expression, termed a cytokine storm. Cytokine storms drive hyperinflammation that can be toxic to organs, and could ultimately lead to multiple organ failure in severe cases of COVID-19. Critically ill COVID-19 patients also display abnormal thrombosis and low platelet counts, suggesting that coagulation processes are critical components in the final stages of COVID-19. In the majority of COVID-19-related deaths, disseminated intravascular coagulation (DIC) occurs. DIC is a condition in which small blood vessels throughout the body are blocked by blood clots.
Platelets are small anucleated megakaryocyte cell fragments generated in bone marrow and the lung, and are essential during thrombus formation. Under normal wound healing events, platelet receptors bind immobilized Von Willebrand factor (VWF) and collagen located in exposed subendothelial matrix, resulting in platelet adhesion. Platelets are then activated to induce conformational changes that lead to the exposure of integrin binding sites necessary for platelet-to-platelet aggregation and platelet plug formation. Additional platelets are attracted through the release of dense platelet granules. Platelet plugs are stabilized through binding fibrinogen.Occurring concomitantly with platelet aggregation, the extrinsic pathway is initiated by tissue factor (TF) to activate the generation of thrombin that is required to form a fibrin mesh. Thrombin is then responsible for activating the intrinsic pathway to facilitate repeating coagulation events to form clots in a positive feedback loop. To prevent thrombosis, fibrinolysis occurs to dissolve blood clots during wound healing, facilitated by plasmin and serpins.
Clinical manifestations of COVID-19 can include thrombocytopenia (low platelet count), accompanied by microvascular thrombosis and increased pathological clotting. Due to the association of abnormal clotting with lethality in severe COVID-19 patients, it is important to understand the underlying mechanisms driving these thrombotic events. To explore this further, we have compiled a list of potential mechanisms and future research directions that could explain how abnormal clotting occurs while platelet count is low during COVID-19 pathogenesis. These ideas are originally published in Xu et al. 2020 and Zaid et al. 2020, with the latter being released prior to peer-review.
1. SARS-CoV-2 could be directly reducing platelet production
To explore this hypothesis, mechanisms of activity used by other coronaviruses are considered. It is reported that infection with coronaviruses SARS-CoV and HCoV-229E cause thrombocytopenia, similar to the thrombocytopenia seen in COVID-19. HCoV-229E functions through the CD13 cell surface receptor. CD13 is expressed on the surface of epithelial cells in the lung and respiratory tract, as well as on activated endothelial cells, lymphocytes, and platelets. After entering bone marrow cells and platelets through CD13, HCoV-229E induces apoptosis and reduces hematopoiesis, resulting in thrombocytopenia.
Due to similarities in antigen characteristics between the SARS-CoV-2, SARS-CoV, and HCoV-229E coronaviruses, along with the comparable presentation of thrombocytopenia, it is speculated that SARS-CoV-2 may be directly inhibiting hematopoiesis in bone marrow (Fig 1). After entering bone marrow and platelet cells through a cell surface receptor, it is speculated that SARS-CoV-2 could be inhibiting cell growth and promoting apoptosis, ultimately leading to decreased platelet production and thrombocytopenia during COVID-19.

Figure 1: SARS-CoV-2 could be directly infecting platelet cells and decreasing platelet production
Future Directions to Consider:
- Does SARS-CoV-2 directly enter bone marrow cells and platelets through a cell surface receptor? If so, is this through ACE2 or another receptor?
- Do hematopoietic cells and platelets undergo apoptosis following SARS-CoV-2 infection?
- Could a cytokine storm destroy or skew differentiation of hematopoietic progenitor cells in the bone marrow, resulting in decreased platelet production? This could be an indirect effect of SARS-CoV-2 infection.
- What happens to COVID-19 outcomes when patients have preexisting low platelet counts due to underlying conditions like cancer and treatment with chemotherapy?
- How is the hematopoietic potential of the lungs impacted in COVID-19? Are the platelets released from megakaryocytes in the lung, rather than in the blood marrow, undergoing the same changes as the bone marrow-released platelets?
2. SARS-CoV-2 could be promoting platelet destruction
Increased levels of immune complexes and autoantibodies are observed in COVID-19, which could be leading to immune-mediated thrombocytopenia. Immune-mediated thrombocytopenia occurs when the immune system specifically targets and destroys platelets. This happens when autoantibodies and immune complexes are deposited on the surface of platelets. These platelets then become targets for immune-mediated damage and destruction (Fig 2). COVID-19 associated immune thrombocytopenic purpura is caused by platelet autoantibodies and can lead to life-threatening bleeding (Lévesque et al, 2020). Molecular mimicry may also be occurring, where antibodies are produced in response to viral infection that specifically target antigens on platelets, ultimately tagging them for destruction.
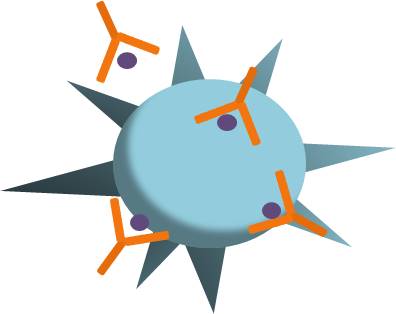
Figure 2: Antigens on platelets may be targeted by the immune system following SARS-CoV-2 infection.
Future Directions to Consider:
- Are immune complexes, autoantibodies, or other antibodies binding to platelets in COVID-19? If so, does this lead to platelet destruction?
- If this is immune-mediated, do immune TTP therapies improve outcomes for COVID-19 patients?
- Is there a correlation of immune-mediated reactions and the level of immune responsiveness to decreased platelet numbers?
3. Platelets may be consumed during COVID-19 thrombus formation
Viral infection in the lungs causes damage in the lung and perhaps other organs. The damaged tissue in turn activates platelets to aggregate and form microthrombi. The increasing number of microthrombi may be consuming circulating platelets (Fig 3).
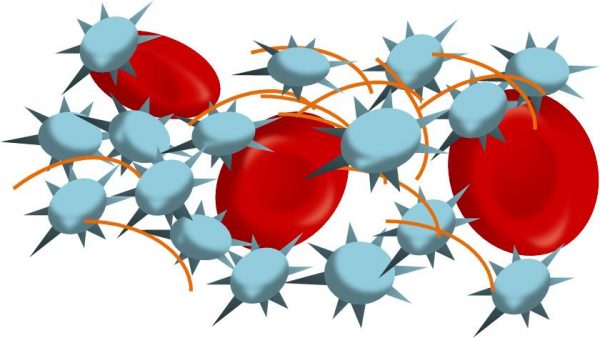
Figure 3: Activated platelets could be aggregating to form clots. These microthrombi are consuming the number of circulating platelets.
Future Directions to Consider:
- Does therapeutic treatment for thrombocytopenia in COVID-19 patients result in improved outcomes?
- Does SARS-CoV-2 infection alter the activation and aggregation of platelets such that platelets are more prone to accumulate into microthombi?
- Does the level of viral load or immune responsiveness correlate with damage and thus platelet consumption?
4. Platelets could be hyper-activated in COVID-19
As platelets are critical for hemostasis, they are also important components of inflammation. To promote inflammation, platelets express inflammatory cytokines, which attract neutrophils into inflamed tissues. A recent study (Zaid et al, 2020) examined blood samples collected from severe COVID-19 patients. These blood samples exhibit low platelet counts and high levels of the inflammation marker lactate dehydrogenase (LDH). Viral RNA was also identified in platelets collected from COVID-19 patients. These platelets, although low in number, were more likely to produce inflammatory cytokines when stimulated by thrombin compared to platelets from nonsevere COVID-19 individuals. The study therefore suggests that platelets are primed to produce inflammatory cytokines following SARS-CoV-2 infection, and are more likely to release extracellular vesicles, which are part of the coagulation and inflammation processes (Fig 4). PKCδ phosphorylation, which is necessary for platelet aggregation and activation, was significantly increased on the platelets from COVID-19 patients following stimulation with thrombin. Under flow conditions, severe COVID-19 platelets adhered to collagen-coated surfaces more efficiently than non-severe and healthy controls (Zaid et al, 2020). The study concludes that platelets, although less in number, are hyper-active and may be contributing to increased inflammation and thrombosis following SARSCoV-2 infection (Fig 4).
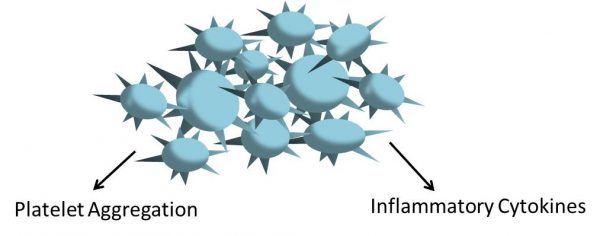
Figure 4: Platelets may become hyper-activated following SARS-CoV-2 infection, leading to increased inflammatory cytokine production and platelet aggregation.
Future Directions to Consider:
- Are platelets from COVID-19 patients better able to activate neutrophils than non-COVID-19 samples? Does this correlate with disease severity?
- How are thrombosis, inflammation, and COVID-19 outcomes affected after blocking platelet activation pathways? Is there a decrease in disease severity or thrombi
formation following anti-platelet therapies or in individuals with underlying defects in platelet activation? Does this correlate with disease severity? - How long does platelet priming last? Is platelet hyper-activity contributing to the spontaneous clotting issues that have been observed in the weeks following mild cases of COVID-19?
- Would the risk for severe COVID-19 complications be higher in people who are already at high risk for thrombosis?
- Are platelets directly infected by SARS-CoV-2, or is viral RNA being transferred to platelets from megakaryocytes, or from some other source?
Referenced Papers
Lévesque V, Millaire E, Corsilli D, Rioux-Massé B, Carrier F. Severe immune thrombocytopenic purpura in critical COVID-19. Int J Hematol. 2020 Jul 1;1-5.
Xu P, Zhou Q, Xu J. Mechanism of thrombocytopenia in COVID-19 patients. 500 Ann Hematol. 2020;99(6):1205-1208.
Zaid, Y. et al. (2020). Platelets Can Contain SARS-Cov-2 RNA And Are Hyperactivated in COVID-19. medRxiv preprint. doi: https://doi.org/10.1101/2020.06.23.20137596.
ML-00-00663Rev01