Emerging Therapeutic Targets for Hereditary Angioedema
Posted on: August 31, 2022
-Contributed by Abi Kasberg, PhD
Hereditary Angioedema (HAE) is a rare, inherited, autosomal dominant disorder that is characterized by fluid accumulation and tissue swelling that can be painful and potentially life-threatening. This form of angioedema is driven by vasculature leakage into surrounding tissues including in the hands, feet, lips, genitals, gastrointestinal tract, or airway. Normal blood flow and lymphatic fluid flow can be impaired in HAE. HAE episodes can occur unexpectedly and at a rate of up to several attacks per month. HAE symptoms can be triggered by or become more severe following surgery, dental procedures, illness, stress, or severe injury. Careful prevention and management of HAE attacks is critically important to providing the best care and quality of life for individuals with HAE.
Type I and Type II HAE is caused by quantitative deficiencies or impaired functional activity of C1 esterase inhibitor (C1-INH), respectively. C1-INH is primarily produced in the liver and circulates in plasma (Ameratunga et al. 2016). As a multifunctional serine protease inhibitor, C1-INH regulates the activity of complement protein C1. C1-INH also inhibits contact activation system (CAS) proteins of the coagulation pathway including factor XIIa (FXIIa) and kallikrein (Fig. 1). When C1-INH activity is lost or impaired, CAS proteins are activated, resulting in the accumulation of bradykinin, a potent vasodilator (Simão and Feener 2017). Excess levels of bradykinin bind bradykinin B2 receptors and activate endothelial cells in a manner that impairs vascular integrity and leads to angioedema (Fig. 1) (Kajdácsi et al. 2014, Simão and Feener 2017).
There is a third type of HAE (HAE-nC1) where C1-INH levels are normal and disease symptoms are driven by other genetic causes. Mutations in FXII, plasminogen, and other unknown genetic causes have been shown to drive HAE-nC1. For the purposes of this article, the term HAE refers to Type I and Type II HAE, where C1-INH deficiencies drive disease pathogenesis.
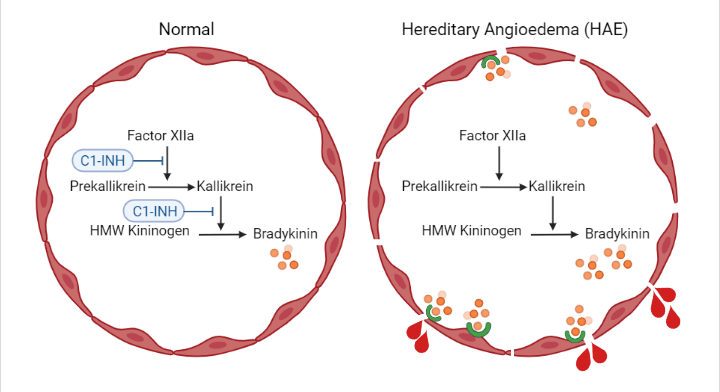
Fig. 1: Hereditary Angioedema (HAE) is caused by the absence or impaired functionality of C1 esterase inhibitor (C1-INH). C1-INH deficiency results in the accumulation of the vasodilator bradykinin. Bradykinin activates vascular endothelial cells and enhances vascular permeability through binding B2 receptors. This leads to angioedema and loss of vascular integrity in HAE. Figure created with BioRender.com
HAE Therapeutics
There are currently three indications to treat HAE. The first is on-demand therapy that is administered following the onset of symptoms. Short-term prophylaxis therapy is a second option that can be used to prevent HAE episodes prior to high-risk procedures, such as before surgery. Finally, long-term prophylaxis therapy is aimed to reduce the overall number of HAE episodes, particularly in patients requiring more assistance in disease control compared to on-demand therapy alone (Nicola et al. 2019).
Several therapeutic directions are currently available to treat HAE, such as C1-INH replacement therapy, attenuated androgenic hormone therapy, antifibrinolytic drugs, and innovative gene therapies. Benefits and risks are associated with each therapeutic option. Androgen therapy can cause liver failure and virilization, along with possible interference with growth and sexual maturation (Nicola et al. 2019, Zuraw 2010a). Antifibrinolytic drugs are preferred over hormone therapy in children and pregnant individuals, however the specific mechanism of antifibrinolytic activity during HAE treatment is not well understood (Zuraw 2010a). Frozen human plasma-derived therapeutics, which are commonly used during C1-INH replacement therapies, can be associated with elevated risks of infectious disease transmission.
To expand upon and improve HAE treatment options, novel HAE treatment strategies are being developed in the fields of gene therapy, molecular inhibitors, and monoclonal antibodies (mAbs). Given the complexity of the CAS cascade that is impacted by C1-INH deficiencies, different therapeutic targets are being investigated to treat HAE.
C1-INH Replacement Therapies
The primary aim of C1-INH replacement therapies is to target the principal cause of the disorder, C1-INH deficiencies. Multiple strategies are being investigated and utilized to improve C1-INH activity and restore C1-INH levels in HAE individuals. Historically, C1-INH levels have been replaced with plasma-derived C1-INH or with recombinant C1-INH. A novel and promising therapeutic approach being developed to treat HAE is through gene therapy targeting C1-INH expression.
C1-INH Gene Therapies
Gene therapies are being developed that utilize adeno-associated viral vectors to express the human SERPING1 gene that encodes the C1-INH protein (Fig. 2). This strategy aims to diminish the need for long-term or recurring clinical treatments by restoring C1-INH levels and ultimately reducing the number and frequency of HAE attacks. Gene therapy candidate ADVM-053 (AAVrh.10hC1EI), from Adverum Biotechnologies, delivers an extra chromosomal copy of SERPING1 in efforts to restore levels of C1-INH (Table 1). In mouse models of HAE, AAVrh.10hC1EI restored C1-INH levels and rescued vascular permeability defects (Qiu et al. 2019).
BioMarin Pharmaceutical is developing a one-time gene therapy treatment with BMN 331 for HAE. BMN 331 is an adeno-associated viral vector that drives expression of human SERPING1 under the control of a liver-selective promoter (Table 1). BMN 331 is currently in clinical trial phase 1/2 for the treatment of HAE with C1-INH deficiency (NCT05121376).
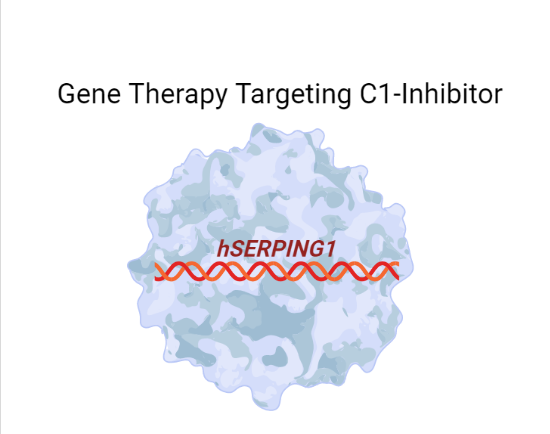
Fig. 2: Adeno-associated viral vector-based gene therapy drives expression of human C1-INH to treat HAE. Figure created with BioRender.com
Plasma-derived C1-INH
C1-INH replacement strategies using plasma-derived C1-INH products (pdC1-INH) have historically been used to treat HAE. Prior to the development of purification technologies to remove bacterial and viral pathogens from donor plasma, there was an elevated risk of transmission of blood-borne pathogens from donor to recipients (Moldovan et al. 2015). Pasteurization and nanofilter techniques are now being employed on currently available pdC1-INH products to remove infectious agents, although there is concern that nonenveloped viruses and other unknown pathogens may contaminate pdC1-INH therapeutics (Moldovan et al. 2015).
Several plasma-derived C1-INH products are available on the market (Table 1). Cinryze® (Shire) is a pdC1-INH nanofiltered concentrate that was the first C1-INH approved by the FDA for treatment in children and adults (Zuraw et al. 2010b). Berinert® (CSL Behring) is a pasteurized pdC1-INH concentrate that is indicated for on-demand treatment (Craig et al. 2011). Haegarda® (CSL Behring) is a pdC1-INH product that is approved by the FDA for use as a self-administered subcutaneous dose of C1-INH concentrate to treat HAE. A phase-3 clinical trial was recently completed for CSL830 (CSL Behring), a pasteurized, nanofiltered, human pdC1-INH preparation (NCT01912456). Study results suggest that prophylactic use of subcutaneously administered CSL830 reduces the frequency of HAE attacks (Longhurst et al. 2017).
Recombinant human C1-INH
Recombinant technology has made it possible to produce human C1-INH without being dependent on human blood. Recombinant C1-INH produced in mammalian cell lines circumvents the potential for blood-borne pathogen transmission while also improving the purity of the final product, independent from human donor supply constraints. There remains the possibility of cross-species viral infection which requires the need for robust viral testing and elimination steps to remove possible viruses from the recombinant production system (Moldovan et al. 2015). Despite the risks, it is generally regarded and supported by the FDA that recombinant products are superior to plasma-derived products in terms of limiting transmission of infectious pathogens (Moldovan et al. 2015).
Ruconest® (Pharming Technologies) is a recombinant human C1-INH (rhC1INH) that is FDA-approved for the treatment of acute HAE attacks (Table 1). Ruconest® is generated in transgenic New Zealand White (NZW) rabbits that express recombinant human C1-INH in mammary gland specific expression vectors. This transgenic platform produced rhC1INH at high expression rates that can scaled up due to the short breeding time of NZW rabbits compared to other mammalian species (Moldovan et al. 2015). Compared to pdC1-INH products Berinert® and Cinryze®. Ruconest® has higher specific activity and purity (Moldovan et al. 2015). rhC1INH has been investigated for use as a prophylaxis treatment of HAE in a phase 2 clinical trial (NCT02247739). Results from this trial suggest that rhC1INH reduces the frequency of HAE attacks (Reshef et al. 2013, Riedl et al. 2017).
Takeda (Shire) has also sponsored and completed a phase 1 clinical study investigating the safety of recombinant human C1-INH, SHP623 (Table 1) (NCT02663687).
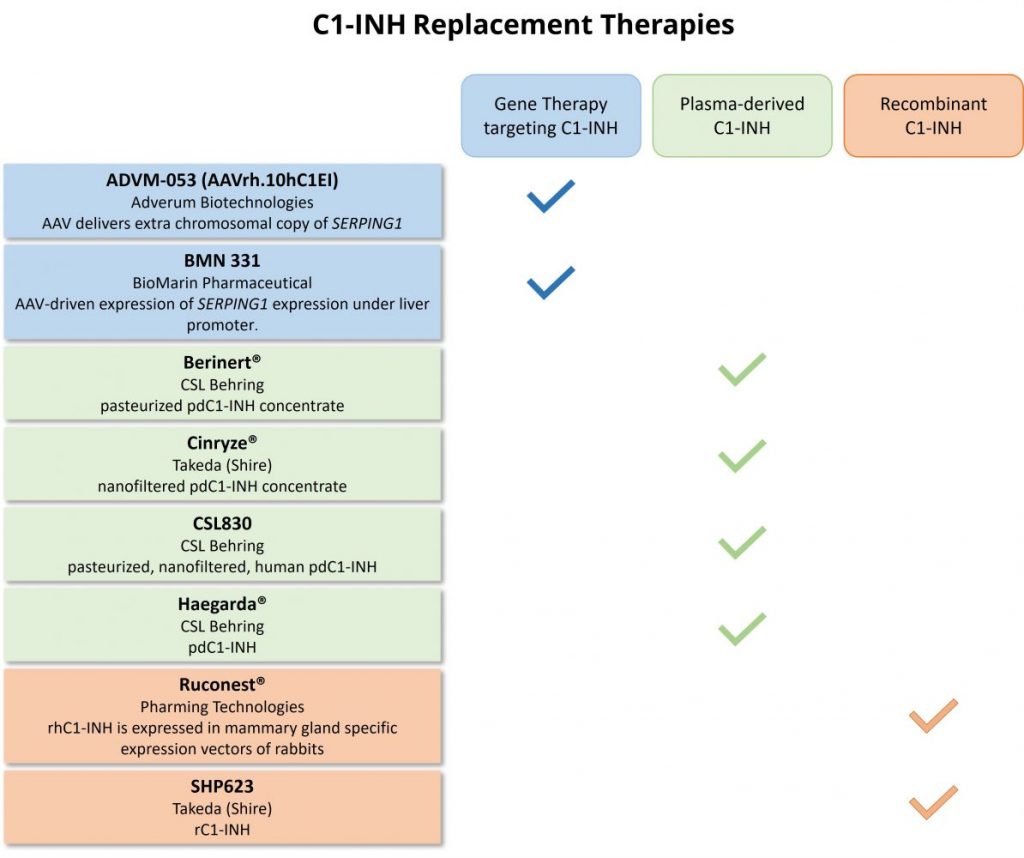
Table 1: Proposed therapeutics that target C1-INH to treat HAE.
HAE Therapies with Non-C1-INH Targets
Many HAE therapeutics are on the market and are in the throngs of research and development that do not target to replace C1-INH expression or activity. Instead, therapeutic strategies are targeting additional components of the CAS pathway in efforts to restore downstream bradykinin to normal levels. Attempts are being developed to inhibit the expression or activity of prekallikrein, kallikrein, Factor XIIa, and bradykinin B2 receptor as treatment options for HAE (Fig. 3). Table 2 provides a summary of the HAE therapeutics available or in development that have non-C1-INH targets.
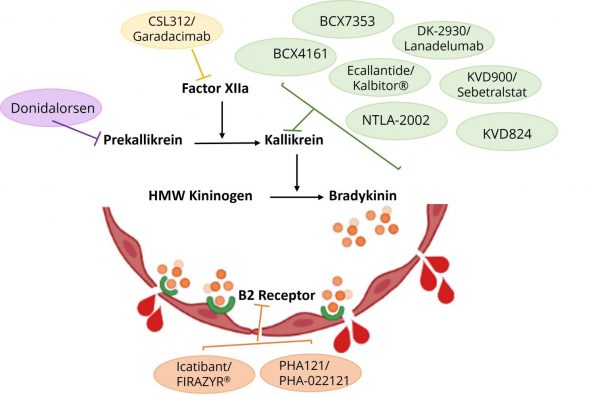
Fig. 3: HAE therapeutics are being pursued and investigated to target proteins of the contact activation system (CAS) pathway in efforts to either prevent the accumulation of bradykinin or inhibit the activation of bradykinin on B2 receptors.
| Drug, Manufacturer | Target | Technology | Mechanism | References |
|
Donidalorsen Ionis Pharmaceuticals |
Prekallikrein | Ligand-conjugated antisense (LICA) chemical technology | Antisense oligonucleotide that reduces the production of prekallikrein |
Fijen et al. 2022 NCT05392114, NCT04030598 |
|
BCX4161 BioCryst Pharmaceuticals |
Kallikrein | Small molecule inhibitor | Oral kallikrein inhibitor | NCT02303626 |
|
BCX7353 BioCryst Pharmaceuticals |
Kallikrein | Small molecule inhibitor | Synthetic, oral plasma kallikrein inhibitor |
Manning et al. 2021 NCT03472040 |
|
DK-2930 / Lanadelumab Takeda (Shire) |
Kallikrein | Recombinant human monoclonal antibody | Monoclonal antibody that inhibits active plasma kallikrein |
Banerji et al. 2018 NCT01923207, NCT02586805 |
|
Ecallantide / Kalbitor® Takeda (Shire) |
Kallikrein | Yeast recombinant technology | Subcutaneous treatment that inhibits plasma kallikrein |
Levy et al. 2010 MacGinnitie et al. 2013 NCT01059526, NCT00456508 |
|
KVD824 KalVista Pharmaceuticals |
Kallikrein | Small molecule protease inhibitor | Oral plasma kallikrein inhibitor |
NCT05055258 NCT05178355 |
|
KVD900 / Sebetralstat KalVista Pharmaceuticals |
Kallikrein | Small molecule protease inhibitor | Oral plasma kallikrein inhibitor |
Duckworth 2022 Maetzel et al. 2022 NCT04349800, NCT04208412 |
|
NTLA-2002 Intellia Therapeutics |
Kallikrein |
Gene therapy, CRISPR/Cas9 gene editing system |
Inactivation of KLKB1 gene reduces plasma kallikrein activity | NCT05120830 |
|
CSL312 / Garadacimab CSL Behring |
Factor XIIa | Recombinant human IgG monoclonal antibody | Factor XIIa antagonist monoclonal antibody (FXIIa mAb) |
NCT04739059 NCT04656418 |
|
Icatibant / FIRAZYR® Takeda (Shire) |
Bradykinin B2 receptor | Synthetic peptidomimetic drug that antagonizes B2 receptors | Bradykinin B2 receptor antagonist blocks bradykinin binding to B2 receptors | Dubois and Cohen 2010 |
|
PHA121 / PHA-022121 Pharvaris |
Bradykinin B2 receptor | Small molecule inhibitor | Oral bradykinin B2 receptor antagonist |
NCT04618211 NCT05047185 |
Table 2: Proposed inhibitor therapeutics that have non-C1-INH targets and utilize distinctive mechanisms to treat HAE.
Summary
There remains an incomplete understanding of the pathogenesis driving HAE, highlighted by the unpredictable occurrence and severity of HAE attacks. As discussed, a variety of therapeutic technologies, modes of delivery, and molecular targets are being utilized to treat HAE and expand HAE research. Medical advances in subcutaneous drug administration and oral medications have enabled patients with HAE to have the flexibility to self-administer treatment at home. Approved HAE treatments do a fair job at preventing or minimizing HAE attacks but fall short of curing or correcting the cause of the disease at its source. C1-INH gene therapy offers encouraging opportunities to correct the causal deficiency that is driving HAE, in hopes of providing a cure and improved lifelong quality of life. Despite these innovations, more needs to be explored and considered regarding the implementation of novel therapies, such as drug cost, efficacy, and administration (Nicola et al. 2019). The recent and upcoming advances in HAE treatments will create a promising menu of treatments options to enable individualized approaches to HAE treatment and long-term disease management.
Further Reading
Ameratunga R, Bartlett A, McCall J, Steele R, Woon ST, Katelaris CH. Hereditary Angioedema as a Metabolic Liver Disorder: Novel Therapeutic Options and Prospects for Cure. Front Immunol. 2016 Nov 30;7:547. doi: 10.3389/fimmu.2016.00547. PMID: 27965672; PMCID: PMC5127832.
Banerji A, Riedl MA, Bernstein JA, Cicardi M, Longhurst HJ, Zuraw BL, Busse PJ, Anderson J, Magerl M, Martinez-Saguer I, Davis-Lorton M, Zanichelli A, Li HH, Craig T, Jacobs J, Johnston DT, Shapiro R, Yang WH, Lumry WR, Manning ME, Schwartz LB, Shennak M, Soteres D, Zaragoza-Urdaz RH, Gierer S, Smith AM, Tachdjian R, Wedner HJ, Hebert J, Rehman SM, Staubach P, Schranz J, Baptista J, Nothaft W, Maurer M; HELP Investigators. Effect of Lanadelumab Compared With Placebo on Prevention of Hereditary Angioedema Attacks: A Randomized Clinical Trial. JAMA. 2018 Nov 27;320(20):2108-2121. doi: 10.1001/jama.2018.16773. Erratum in: JAMA. 2019 Apr 23;321(16):1636. PMID: 30480729; PMCID: PMC6583584.
Craig TJ, Bewtra AK, Bahna SL, Hurewitz D, Schneider LC, Levy RJ, Moy JN, Offenberger J, Jacobson KW, Yang WH, Eidelman F, Janss G, Packer FR, Rojavin MA, Machnig T, Keinecke HO, Wasserman RL. C1 esterase inhibitor concentrate in 1085 Hereditary Angioedema attacks–final results of the I.M.P.A.C.T.2 study. Allergy. 2011 Dec;66(12):1604-11. doi: 10.1111/j.1398-9995.2011.02702.x. Epub 2011 Sep 2. PMID: 21884533.
Dubois EA, Cohen AF. Icatibant. Br J Clin Pharmacol. 2010 May;69(5):425-6. doi: 10.1111/j.1365-2125.2010.03642.x. PMID: 20573077; PMCID: PMC2856042.
Duckworth EJ, Murugesan N, Li L, Rushbrooke LJ, Lee DK, De Donatis GM, Maetzel A, Yea CM, Hampton SL, Feener EP. Pharmacological suppression of the kallikrein kinin system with KVD900: An orally available plasma kallikrein inhibitor for the on-demand treatment of hereditary angioedema. Clin Exp Allergy. 2022 Mar 12. doi: 10.1111/cea.14122. Epub ahead of print. PMID: 35278245.
Fijen LM, Riedl MA, Bordone L, Bernstein JA, Raasch J, Tachdjian R, Craig T, Lumry WR, Manning ME, Alexander VJ, Newman KB, Revenko A, Baker BF, Nanavati C, MacLeod AR, Schneider E, Cohn DM. Inhibition of Prekallikrein for Hereditary Angioedema. N Engl J Med. 2022 Mar 17;386(11):1026-1033. doi: 10.1056/NEJMoa2109329. PMID: 35294812.
Kajdácsi E, Jani PK, Csuka D, Varga LÁ, Prohászka Z, Farkas H, Cervenak L. Endothelial cell activation during edematous attacks of hereditary angioedema types I and II. J Allergy Clin Immunol. 2014 Jun;133(6):1686-91. doi: 10.1016/j.jaci.2013.12.1072. Epub 2014 Feb 9. PMID: 24522092.
Levy RJ, Lumry WR, McNeil DL, Li HH, Campion M, Horn PT, Pullman WE. EDEMA4: a phase 3, double-blind study of subcutaneous ecallantide treatment for acute attacks of hereditary angioedema. Ann Allergy Asthma Immunol. 2010 Jun;104(6):523-9. doi: 10.1016/j.anai.2010.04.012. PMID: 20568386.
Longhurst H, Cicardi M, Craig T, Bork K, Grattan C, Baker J, Li HH, Reshef A, Bonner J, Bernstein JA, Anderson J, Lumry WR, Farkas H, Katelaris CH, Sussman GL, Jacobs J, Riedl M, Manning ME, Hebert J, Keith PK, Kivity S, Neri S, Levy DS, Baeza ML, Nathan R, Schwartz LB, Caballero T, Yang W, Crisan I, Hernandez MD, Hussain I, Tarzi M, Ritchie B, Králíčková P, Guilarte M, Rehman SM, Banerji A, Gower RG, Bensen-Kennedy D, Edelman J, Feuersenger H, Lawo JP, Machnig T, Pawaskar D, Pragst I, Zuraw BL; COMPACT Investigators. Prevention of Hereditary Angioedema Attacks with a Subcutaneous C1 Inhibitor. N Engl J Med. 2017 Mar 23;376(12):1131-1140. doi: 10.1056/NEJMoa1613627. PMID: 28328347.
MacGinnitie AJ, Davis-Lorton M, Stolz LE, Tachdjian R. Use of ecallantide in pediatric hereditary angioedema. Pediatrics. 2013 Aug;132(2):e490-7. doi: 10.1542/peds.2013-0646. Epub 2013 Jul 22. PMID: 23878046.
Maetzel A, Smith MD, Duckworth EJ, Hampton SL, De Donatis GM, Murugesan N, Rushbrooke LJ, Li L, Francombe D, Feener EP, Yea CM. KVD900, an oral on-demand treatment for hereditary angioedema: Phase 1 study results. J Allergy Clin Immunol. 2022 Jun;149(6):2034-2042. doi: 10.1016/j.jaci.2021.10.038. Epub 2022 Jan 24. PMID: 35086692.
Manning ME, Kashkin JM. Berotralstat (BCX7353) is a novel oral prophylactic treatment for hereditary angioedema: Review of phase II and III studies. Allergy Asthma Proc. 2021 Jul 14;42(4):274-282. doi: 10.2500/aap.2021.42.210034. Epub 2021 Jun 14. PMID: 34127176.
Moldovan D, Bernstein JA, Cicardi M. Recombinant replacement therapy for hereditary angioedema due to C1 inhibitor deficiency. Immunotherapy. 2015;7(7):739-52. doi: 10.2217/imt.15.44. Epub 2015 Aug 7. Erratum in: Immunotherapy. 2015;7(11):1229-131. PMID: 26250409.
Nicola S, Rolla G, Brussino L. Breakthroughs in hereditary angioedema management: a systematic review of approved drugs and those under research. Drugs Context. 2019 Oct 2;8:212605. doi: 10.7573/dic.212605. Erratum in: Drugs Context. 2019 Dec 30;8: PMID: 31645881; PMCID: PMC6788388.
Qiu T, Chiuchiolo MJ, Whaley AS, Russo AR, Sondhi D, Kaminsky SM, Crystal RG, Pagovich OE. Gene therapy for C1 esterase inhibitor deficiency in a Murine Model of Hereditary angioedema. Allergy. 2019 Jun;74(6):1081-1089. doi: 10.1111/all.13582. Epub 2019 Mar 19. PMID: 30059156; PMCID: PMC6752709.
Reshef A, Moldovan D, Obtulowicz K, Leibovich I, Mihaly E, Visscher S, Relan A. Recombinant human C1 inhibitor for the prophylaxis of hereditary angioedema attacks: a pilot study. Allergy. 2013 Jan;68(1):118-24. doi: 10.1111/all.12060. Epub 2012 Nov 5. PMID: 23121116.
Riedl MA, Grivcheva-Panovska V, Moldovan D, Baker J, Yang WH, Giannetti BM, Reshef A, Andrejevic S, Lockey RF, Hakl R, Kivity S, Harper JR, Relan A, Cicardi M. Recombinant human C1 esterase inhibitor for prophylaxis of hereditary angio-oedema: a phase 2, multicentre, randomised, double-blind, placebo-controlled crossover trial. Lancet. 2017 Sep 30;390(10102):1595-1602. doi: 10.1016/S0140-6736(17)31963-3. Epub 2017 Jul 25. PMID: 28754491.
Simão F, Feener EP. The Effects of the Contact Activation System on Hemorrhage. Front Med (Lausanne). 2017 Jul 31;4:121. doi: 10.3389/fmed.2017.00121. PMID: 28824910; PMCID: PMC5534673.
Zuraw BL. HAE therapies: past present and future. Allergy Asthma Clin Immunol. 2010a Jul 28;6(1):23. doi: 10.1186/1710-1492-6-23. PMID: 20667126; PMCID: PMC2921104.
Zuraw BL, Busse PJ, White M, Jacobs J, Lumry W, Baker J, Craig T, Grant JA, Hurewitz D, Bielory L, Cartwright WE, Koleilat M, Ryan W, Schaefer O, Manning M, Patel P, Bernstein JA, Friedman RA, Wilkinson R, Tanner D, Kohler G, Gunther G, Levy R, McClellan J, Redhead J, Guss D, Heyman E, Blumenstein BA, Kalfus I, Frank MM. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010b Aug 5;363(6):513-22. doi: 10.1056/NEJMoa0805538. PMID: 20818886.
ML-00-00954Rev01