Activated Protein C (APC) is a regulator of the coagulation cascade, by specifically inactivating factors Va and VIIIa, in the presence of phospholipids and calcium.
In most of the cases (more than 90%), Activated Protein C Resistance (APCR) phenotype is caused by a Factor V gene mutation (Factor V Leiden). The mutation, located on Factor V exon 10 (1691 G –> A), of arginine to glutamine on position 506, renders Factor Va resistant to the cleavage by Activated Protein C. This genetic anomaly can be evidenced with a clotting method performed in the presence or the absence of Activated Protein C.
Activated Protein C Resistance is tested by using a clotting method performed with or without Activated Protein C.
Introduction
Activated protein C (APC) is a key anticoagulant enzyme needed for the proper down-regulation of blood coagulation. A poor anticoagulant response to APC, denoted APC resistance, is a recently described blood defect found to be a major risk factor for venous thromboembolism in Western societies. At least 90% of cases with the APC resistance phenotype can be explained by a point mutation in the coagulation factor V gene, changing arginine 506 in the factor V molecule to glutamine. After activation, mutated factor V (denoted FV:Q506 or Factor V Leiden) is partially resistant to inactivation by APC, which allows for longer duration of thrombin generation and may lead to a hypercoagulable state. APC resistance due to the presence of the FV:Q506 allele is inherited as an autosomal dominant trait and has a prevalence of 2-13% in the general population. Frequencies of APC resistance among patients with venous thrombosis, depending on the selection criteria, range from 20-60%. The high prevalence of APC resistance, combined with the availability of simple blood tests by which it is detected, raises the question of whether general screening for APC resistance should be performed in conjunction with circumstantial risk factors for thrombosis such as surgery, pregnancy and oral contraceptives.
Virchows triad for venous thrombosis
Venous thromboembolism
The formation of an obstructive mass of clotted blood in the venous part of the circulatory system is known as venous thrombosis. The mass itself is called a thrombus and is composed of platelets, blood cells and fibrin. A thrombus which breaks loose and is carried away with the bloodstream is called an embolus. When caught in the blood vessels of the lung it may develop into pulmonary embolism, the most feared complication of venous thrombosis.
Major health problem in Western countries
Venous thromboembolism is a major health problem in Western societies, constituting the third most common cardiovascular disease after acute ischemic heart disease and stroke. The incidence has increased steadily in recent centuries, perhaps due to longer life-spans and the adoption of more sedentary habits. In the USA, venous thromboembolism accounts for more than 250,000 hospitalizations a year, corresponding to an incidence of about one per 1,000 individuals. The annual death rate due to pulmonary embolism is estimated to be 50,000.
Thrombogenic risk factors
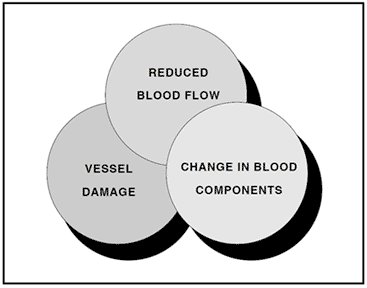
The three primary risk factors for venous thrombosis, as described by Virchow more than a century ago, are related to a reduced blood flow, vessel wall damage, and a change in blood components. Any one of these risk factors potentiates the other and may create a condition known as a hypercoagulable state. In principle, this condition can be viewed as a biochemical imbalance in the blood that favors blood coagulation and the formation of a blood clot.
Risk factors for hypercoagulability and venous thromboembolism can be either acquired or inherited. Acquired risk factors such as surgery, pregnancy, oral contraceptives or immobilization are found in a large portion of patients. In addition, many patients also have a genetically determined tendency to thrombosis, as indicated by the fact that up to 40% of patients referred to a specialist laboratory have a family history of thrombosis. Until recently, three gene disorders had been identified associated with a clear increase in the risk for venous thromboembolism. These hereditary disorders were usually found in only a few percent of thrombosis-prone families through a symptomatic patient with a deficiency of either antithrombin, protein C or protein S. Thus, in vast majority of thrombotic patients no genetic risk factor could be identified.
Novel defect in the protein C anticoagulant pathway
The diagnostic situation for familial thrombosis improved dramatically in 1993 with the discovery of a novel defect in the protein C anticoagulant pathway.Based on the hypothesis that a poor anticoagulant response to activated protein C (APC) might predispose to thrombosis, a Swedish research-team led by Björn Dahlbäck, investigated the activity of exogenously added APC in an APTT-based assay. In a normal response, the addition of APC to plasma induces a prolonged clotting time. This occurs because APC cleaves and inactivates two critical coagulation proteins, factors Va and VIIIa, resulting in a reduced rate of thrombin generation. However, when the assay was run on plasma from a middle-aged man suffering from recurrent episodes of venous thrombosis, the result showed a much shorter prolongation of the clotting time than expected. Several of the man’s relatives demonstrated the same phenotype, characterized by a poor anticoagulant response to APC. Family studies suggested that this blood disorder, denoted APC resistance, was inherited as an autosomal dominant trait. Subsequent investigations found that APC resistance was present in up to 60% of cases with a family history of venous thrombosis and that it was highly prevalent in the general population (1-7%). These results proved APC resistance to be the most common inherited defect associated with thrombosis, being larger than the sum of all other previously identified genetic risk factors.
Mutation in the factor V gene explains APC resistance
The search for the molecular mechanism of APC resistance led to the isolation of a protein from normal plasma, which was able to correct APC resistant plasma in a dose dependent manner. The protein was identified as factor V, suggesting that APC resistance was caused by a genetic defect in the factor V gene. Other studies reached the same conclusion and a point mutation that predicts the replacement of arginine at position 506 in the factor V molecule with glutamine was soon identified. At least 90% of APC resistant cases are explained by this mutation, denoted FV:Q506 or Factor V Leiden.
Diagnostic breakthrough in thrombophilia
The discovery of APC resistance and the identification of the FV:Q506 mutation as its main cause means that a genetic explanation can be found almost as often as nongenetic risk factors in patients with venous thrombosis. A consequence of this diagnostic breakthrough has been a conceptual change in how thrombotic disease is viewed. In this monograph we attempt to summarize the knowledge gained during the last three years about APC resistance and to describe the major tests for its phenotype and genotype. It is our hope that the use of these tests will help establish guidelines for therapy and prophylaxis and lead to reduced morbidity and mortality in thrombosis-prone patients.
Biochemistry of APC Resistance
Initiation and regulation of blood coagulation
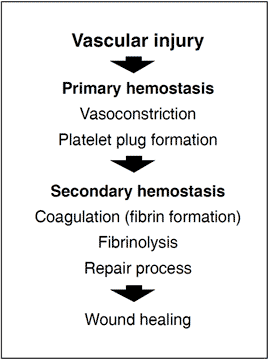
In order to prevent dangerous blood loss following vascular injury, the hemostatic system is called into action. Within seconds of injury the damaged vessel contracts and circulating, disc-shaped cell components called thrombocytes or platelets are activated and start to adhere to the site of injury. The activated platelets then aggregate to form a loose plug that reduces or temporarily stops the bleeding. The activated platelets also release a large number of molecules that accelerate platelet plug formation and begin the process of wound healing. Blood coagulation is triggered simultaneously with these events. This major biochemical process takes place at the surface of negatively charged phospholipid membranes provided by activated platelets and damaged cells. It results in the localized and timely formation of a fibrin matrix that stabilizes the platelet plug and seals the bleeding vessel. The fibrin clot is eventually lysed by the fibrinolytic system at the completion of the healing process.
The coagulation cascade
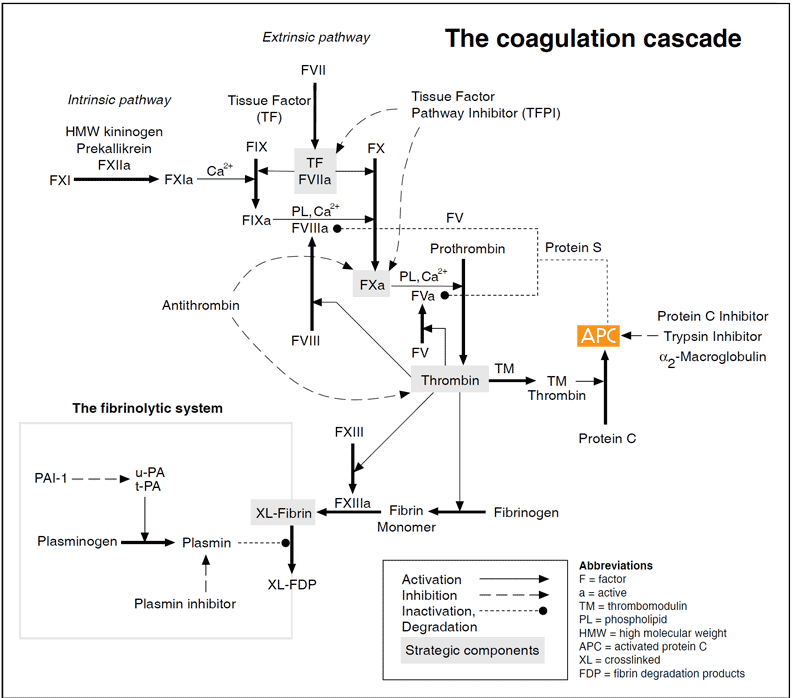
Vascular damage initiates the coagulation cascade resulting in the explosive generation of thrombin at the site of injury. Thrombin catalyzes the conversion of fibrinogen to an insoluble fibrin (clot) matrix, in the presence of factor XIIIa and calcium ions. Critical reactions are closely checked and localized by circulating anticoagulants, such as APC, TFPI and antithrombin. Fibrinolysis is initiated when fibrin is formed and eventually dissolves the clot. Inappropriate activation of blood coagulation and/or depressed fibrinolytic activity may lead to the formation of a thrombus. In contrast, a defect or deficiency in the coagulation process and/or accelerated fibrinolysis is associated with a bleeding tendency. The cascade scheme is organized into the intrinsic (factors XII, XI, IX, VIII, prekallikrein, HMW kininogen), extrinsic (tissue factor, factor VII) and common pathway (factors V, X, XIII, prothrombin, fibrinogen). The extrinsic pathway is initiated when blood is exposed to tissue factor released from damaged endothelium. The intrinsic pathway is initiated by the activation of factor XII involving “contact factors” on negatively-charged surfaces, such as glass or kaolin in vitro. Feedback activations of factors V, VII and VIII by factor Xa and the activation of factor XI by thrombin are not shown.
Although the cascade/waterfall model of blood coagulation has been modified during recent years it is still largely valid. The scheme involves a series of proteolytic reactions, in which inactive coagulation factors in a precursor or zymogen form are activated by one or more cleavages. Several of the activated coagulation proteases form complexes with their specific cofactors on the phospholipid surface, amplifying their activation of subsequent zymogens.
There are two activation pathways in the coagulation cascade, the intrinsic and the extrinsic pathway. The intrinsic pathway involves components intrinsic to whole blood, whereas the extrinsic pathway includes an extrinsic (subendothelial) activating component called tissue factor. Both pathways involve a number of plasma proteins. Most of the coagulation factors are zymogens of trypsin-like serine proteases that cleave arginyl peptide bonds with high specificity. Several proteins, including factors II (prothrombin), VII, IX and X, protein C and protein S, are subjected to vitamin K-dependent g-carboxylation of glutamic acid residues during their synthesis in the liver. This unique amino acid modification allows the proteins to bind calcium ions necessary for phospholipid binding and thereby to participate efficiently in multimolecular complexes in the coagulation cascade. Inhibition of g-carboxylation using vitamin-K-antagonistic drugs such as warfarin is a commonly used approach for anticoagulant treatment.
Initiation of blood coagulation
The extrinsic pathway is now accepted as the major activation route for blood coagulation in vivo. It becomes activated when disrupted tissue and activated monocytes exposes tissue factor to the bloodstream. Tissue factor forms a complex with factor VII, which becomes activated and then activates factors IX and X. The intrinsic pathway is initiated by the exposure of ‘contact’ factors in plasma (i.e. factor XII, HMW kininogen and prekallikrein) to a negatively charged surface, such as connective tissue in vivo or glass in a test tube. The two pathways converge on factor X to a common pathway, leading to the conversion of prothrombin into the key coagulation enzyme, thrombin.
Blood coagulation factors
| Factor | Synonym | M.W. [kDa] | Plasma level [mg/ml] | Function |
|---|---|---|---|---|
| I | Fibrinogen | 340 | 3000 | Structural |
| II | Prothrombin* | 72 | 100 | Protease zymogen |
| III | Tissue factor | 37 | – | Cofactor/initiator |
| IV | Calcium | – | – | – |
| V | Proaccelerin | 330 | 10 | Cofactor precursor |
| VI | – | – | – | – |
| VII | Proconvertin* | 55 | 0.5 | Protease zymogen |
| VIII | Antihemophilic factor | 330 | 0.1 | Cofactor precursor |
| IX | Christmas factor* | 55 | 5 | Protease zymogen |
| X | Stuart-Prower factor* | 55 | 10 | Protease zymogen |
| XI | Thromboplastin antecedent | 160 | 5 | Protease zymogen |
| XII | Hageman factor | 80 | 30 | Protease zymogen |
| XIII | Fibrin-stabilizing factor | 320 | 30 | Protransglutaminase |
| – | Prekallikrein | 85 | 40 | Protease zymogen |
| – | HMW kininogen | 120 | 80 | Cofactor/activation |
| – | Von Willebrand factor | >1500 | 10 | Adhesion, carrier protein |
| 220 kDa subunits |
Regulatory proteins of blood coagulation and fibrinolysis
| Name | M.W. [kDa] | Plasma level [mg/ml] | Function |
|---|---|---|---|
| Tissue factor pathway inhibitor | 40 | 0.1 | Protease inhibitor |
| Antithrombin | 58 | 150 | Protease inhibitor |
| Heparin cofactor II | 66 | 90 | Protease inhibitor |
| Protein C* | 62 | 4 | Protease zymogen |
| Protein S* | 78 | 20 | Cofactor |
| Thrombomodulin | 60 | – | Receptor |
| Protein C inhibitor | 57 | 5 | Protease inhibitor |
| Plasminogen | 92 | 200 | Protease zymogen |
| t-PA | 70 | 0.005 | Protease |
| u-PA | 54 | 0.008 | Protease |
| PAI-1 | 52 | 0.02 | Protease inhibitor |
| PAI-2 | 47 | <0.005 | Protease inhibitor |
| Plasmin Inhibitor | 70 | 70 | Protease inhibitor |
Thrombin escaping from a site of vascular injury binds to its receptor thrombomodulin (TM) on the intact cell surface. As a result,thrombin loses its procoagulant properties and instead becomes a potent activator of protein C. Activated protein C (APC) functions as a circulating anticoagulant, which specifically degrades and inactivates the phospholipid-bound factors Va and VIIIa. This effectively down-regulates the coagulation cascade and limits clot formation to sites of vascular injury. The activity of APC is potentiated by two cofactors, protein S and native (non-activated) factor V. Protein S functions as a cofactor in the degradation of factor Va and VIIIa. Native factor V acts in synergy with protein S as a cofactor in the degradation of factor VIIIa. Thus, factor V has dual roles: one as anticoagulant in its native form and the other as an procoagulant after its activation. APC is slowly neutralized by circulating inhibitors. Thrombin bound to TM is eventually inhibited by antithrombin or removed through endocytosis of the thrombin/TM complex.
Symbols: T= thrombin, PC= protein C, PS= protein S.
The serine protease thrombin converts circulating fibrinogen into clot-forming fibrin molecules and activates the transglutaminase, factor XIII, which stabilizes the fibrin matrix through covalent cross-linking. Thrombin also stimulates cellular hemostasis and coagulation through positive feedback, by activating platelets and the two circulating non-enzymatic cofactor proteins, factor V and factor VIII. All these feedback activations by thrombin lead to an explosive amplification of the coagulation cascade and rapid clot formation.
Thrombin regulation
It is evident that the autocatalytic nature of thrombin could clot the blood content of a person within minutes if uncontrolled. In humans, the necessary control involves two aspects, i.e. inhibition of thrombin already formed and prevention of further thrombin generation. Direct thrombin inhibition is provided primarily by circulating serine protease inhibitor, antithrombin, whereas the crucial prevention of thrombin generation is provided indirectly by thrombin itself. This self-regulating function of thrombin is expressed in its binding to thrombomodulin, a specific, high-affinity receptor protein located on undamaged (intact) endothelium.
The protein C anticoagulant pathway
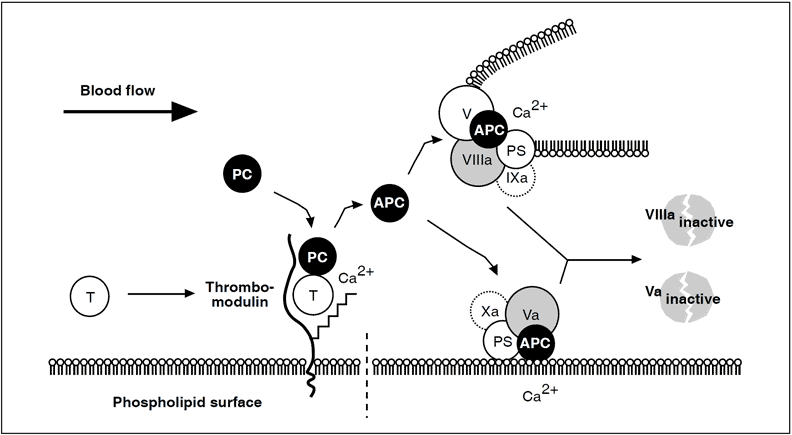
On binding to thrombomodulin, thrombin loses all its procoagulant properties. Instead, it becomes a potent activator of protein C, the ‘prima ballerina’ of the protein C embolic disorder observed in infants with severe protein C or protein S deficiency.
Natural substrates of APC: factors Va and VIIIa
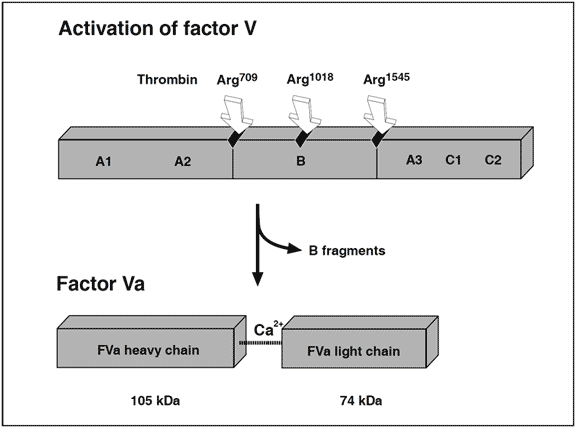
Factors V and VIII are two large, relatively unstable, plasma proteins of about 330 kDa, with similar structure and function. Factor V is an essential component for the rapid conversion of prothrombin to thrombin, whereas factor VIII is needed to accelerate the activation of factor X to factor Xa. The essential role of these non-enzymatic cofactor proteins in hemostasis is evidenced by the severe bleeding tendency associated with their deficiency. Both factors are synthesized mainly in the liver and circulate in plasma as inactive molecules with little or no procoagulant activity. A unique feature of factor VIII is that it circulates in a stabilizing, non-covalent complex with the von Willebrand factor, an adhesive protein that is important for the proper function of platelets. The plasma concentration of factor V is about 10 mg/ml, which is up to a 100-fold higher than that of factor VIII (0.1-0.2 mg/ml). About 20% of the total amount of factor V in blood is synthesized by megacaryocytes and stored in platelets. This stored form of factor V is released in conjunction with platelet activation and has an important role in normal hemostasis. The genes for factor V and VIII are located on chromosomes 1 and X respectively, coding for mature, singlechain proteins of roughly 2200 amino acids. Prior to secretion into the bloodstream, the factor VIII molecule is processed to a calcium ion-linked heterodimer, whereas factor V circulates as a single-chain protein. Computer- aided comparison of the primary amino acid sequence of factors V and VIII reveals a high degree of homology, with an overall identity of about 30%. Both proteins contain several types of similar internal repeats, termed A1-A2-B-A3-C1-C2.
Proteolytic activation of factor V and factor VIII
Factors V and VIII are activated through limited proteolysis by thrombin or factor Xa. During its activation, factor VIII is released from the protective influence of the von Willebrand factor and converted to a calcium ionanticoagulant pathway. The activated protein C (abbreviated APC) is a serine protease that rapidly downregulates thrombin generation, by cleaving and inactivating the phospholipid-bound activated forms of coagulation factors V and VIII (factor Va and factor VIIIa). APC in turn is only slowly neutralized by three inhibitors, protein C inhibitor, trypsin inhibitor and a2-Macroglobulin. The relatively long half-life of APC in vivo (15-20 minutes) is a prerequisite for its function as a circulating anticoagulant.
Protein S
The anticoagulant activity of APC is potentiated and supported by protein S, a non-enzymatic plasma protein. Its mechanism of action is not yet completely understood, but protein S has been shown to promote the binding of APC to phospholipid surfaces, and to remove the factors Xa and IXa-mediated APC protection of factors Va and VIIIa respectively. Protein S has also been reported to stimulate the inactivation of factor Va 20-fold by specific acceleration of the cleavage at Arg306, one of the three cleavage sites for factor Va inactivation. Finally, it may have an APC-independent anticoagulant activity, by inhibiting prothrombin activation through direct interaction with factor Va and factor Xa. About 60% of the protein S in plasma is bound to C4bBP, a regulatory protein of the classic complement system and is not active as APC cofactor.
Anticoagulant role of factor V
Recently, it has been found that non-activated factor V in synergy with protein S functions as an APC cofactor in the degradation of factor VIIIa. This has been confirmed in other studies, although the view of intact factor V as an anticoagulant APC cofactor has recently been challenged. Instead, it has been suggested that the central Bdomain released on activation of factor V expresses the APC cofactor activity. The in vivo relevance of these findings awaits further investigation. Taken as a whole the protein C pathway constitutes an ingenious mechanism by which procoagulant thrombin attains anticoagulant properties in the absence of vascular injury. The physiological importance of this mechanism is demonstrated clinically by the massive thrombo-dependent trimer (A1, A2 and A3-C1-C2). The active factor V molecule is a dimer that consists of a heavy chain (A1-A2), non-covalently linked via calcium ions to a light chain (A3-C1-C2). The activated factors V and VIII bind to negatively charged phospholipid in the presence of calcium and serve as cofactors/receptors for factors IXa and Xa respectively. The importance of these multimolecular complex assemblies, better known as the tenase and prothrombinase complexes, is evidenced by the over 100,000-fold increase in the combined rate of activation of factor X and prothrombin when compared to the activation catalyzed by their respective enzyme alone.
APC inactivation of factors Va and VIIIa
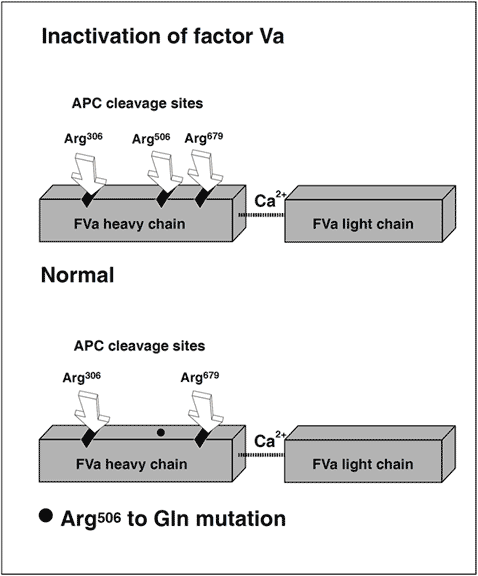
The native form of circulating factors V and VIII are poor substrates for APC. In contrast, APC effectively cleaves and inactivates the phospholipid-bound activated form of these proteins. The inactivation of factor Va takes place through the APC-mediated cleavage in the heavy chain of the molecule of three peptide bonds at Arg506, Arg306 and Arg679. Cleavage at Arg506 is needed for the efficient exposure of the cleavage sites at Arg306 and Arg679. The lipid-dependent cleavage at Arg306 appears to be the major inactivating cleavage site and results in a loss of about 80% cofactor activity, whereas cleavage at Arg679 is lipid-independent and is responsible for the loss of the remaining cofactor activity. Potential structural differences between platelet factor Va and plasma factor Va may influence the extent to which the cofactor is cleaved initially at Arg306. APC inactivates factor VIIIa by cleavages at Arg336, Arg562 and Arg734. The main loss of factor VIIIa cofactor activity is associated with the cleavage at Arg562.
Molecular explanation of APC resistance
The initial observation that normal factor V mixed with APC resistant plasma was able to correct the APC response in a dose-dependent manner, suggested to several independent research groups that APC resistance was due to a defect in the factor V molecule. However, the precise molecular explanation was discovered first by a Dutch group led by R. Bertina.The APC resistance phenotype in this seminal study was linked to a single-point mutation in the factor V gene, which substitutes G (codon CGA) with A (codon CAA) at nucleotide 1691 in exon 10. This mutation replaces Arg (R) with Gln (Q) at position 506 in the factor V molecule, thus modifying one of the three APC cleavage sites. The mutant factor V molecule (abbreviated FV:Q506) expresses normal procoagulant activity when activated by thrombin or factor Xa, although its rate of inactivation is about 10-fold slower than that of normal factor Va. This “resistance” to degradation by APC allows for a longer duration of thrombin generation, which may be reflected by increased levels of coagulation activation markers such as prothrombin fragment 1+2, thrombin-antithrombin (TAT) complex and D-dimer. Recent data also suggest that a reduced ability to slow down thrombin generation may stabilize a blood clot by weakening the profibrinolytic effect of APC.An antifibrinolytic mechanism could thus be an additional factor contributing to the prothrombotic tendency observed in APC resistant patients. The fact that mutant FVa:Q506 can still be inactivated by APC cleavage at Arg306 and Arg679, might account for the relatively mild hypercoagulable state observed in APC-resistant individuals and help explain why additional genetic and/or acquired risk factors are required for thrombosis to develop.
Heterogeneous phenotype
Most cases of inherited APC resistance are caused by the factor V mutation. However, as seen under “normals”, the APC resistance phenotype is clearly heterogeneous, being influenced by other factors as well. Several reports have shown that about 10% (range 4-20%) of APC resistant cases among Caucasians, do not involve the FV:Q506 mutation. The cause of this type of APC resistance is not known but may be the result of acquired APC resistance or due to other genetic defects. Analogous to the FV:Q506 mutation, APC resistance could be explained by mutations at the APC cleavage sites of factor VIII, for example a mutation at Arg336 or Arg562. Although, to date no such mutations have been found.
Summary
Protein C is a vitamin-K-dependent plasma proenzyme of a serine protease that plays a key role in the down-regulation of blood coagulation. It is activated in vivo by the thrombin-thrombomodulin complex on the surface of intact endothelial cells. Activated protein C (APC) functions as a circulating anticoagulant through proteolytic cleavage and inactivation of two critical, phospholipid-bound coagulation proteins, factors Va and VIIIa. The cleavages occur at three sites in the heavy chain of each proteins. The anticoagulant activity of APC is potentiated by protein S and non-activated factor V. APC resistance is mainly due to a point mutation (G to A at nucleotide 1691) in the factor V gene that predicts replacement of arginine at position 506 by glutamine in the factor V molecule. The mutation destroys one of the three APC cleavage sites, rendering the activated and procoagulant factor Va:Q506 partially resistant to APC-mediated degradation. About 90% of APC resistant cases can be explained by the factor V mutation.
Clinical Aspects of APC Resistance
Hypercoagulable state and thrombophilia
A hypercoagulable state is a condition that favours coagulation, as recognized by increased thrombin generation. Hypercoagulability can be due to a number of factors, which can either be inherited (primary) or acquired (secondary).
Thrombophilia is the clinical term for a hypercoagulable state that causes an increased tendency to thrombosis. Several genes have been implicated with inherited thrombophilia, although only factor V (APC resistance), antithrombin, protein C and protein S have been clearly linked to an increased risk of venous thromboembolism. Of these, APC resistance is the most common, both among patients and in the general population.
Causes of hypercoagulability
Primary hypercoagulable states
- Inherited thrombophilia
Secondary hypercoagulable states
- Advanced age
- Heart disease
- Immobility
- Lupus anticoagulants
- Malignancy
- Obesity
- Oral contraceptives
- Pregnancy
- Trauma and surgery
- Varicose veins
Causes of inherited thrombophilia
Established
- APC resistance (factor V:Q506)
- Antithrombin deficiency
- Protein C deficiency
- Protein S deficiency
Non-established
- Dysfibrinogenemia
- Plasminogen deficiency
- Elevated PAI-1
- Heparin Cofactor II deficiency
- Factor XII deficiency
- Hyperhomocysteinemia
Diagnosing APC resistance
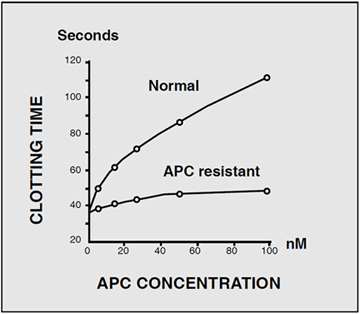
The development of a simple, APTT-based assay that measures the anticoagulant response in plasma to added purified APC, facilitated the characterization of the APC resistance phenotype. In the classic test kit, Coatest® APC™ Resistance, two APTT reactions are performed, one in the presence of a carefully-defined quantity of APC and the other in its absence. The relationship between the two clotting times is expressed as a ratio, called the APC ratio. Healthy individuals have an APC ratio in the range, whereas APC-resistant individuals are recognized by an APC ratio below or equal to about 2. The precise cut-off for a diagnosis may vary slightly depending on the instrument type used as well as the individual condition of the instrument.
Prevalences of inherited thrombophilia in various populations
| Genetic defect | General population | All VT cases | Familial VT cases | No. of mutations |
|---|---|---|---|---|
| APC resistance | 5% | 20% | 50% | 1 |
| Antithrombin deficiency | 0.1% | 1% | 4% | >79 |
| Protein C deficiency | 0.2% | 3% | 5% | >160 |
| Protein S deficiency | n.d. | 2% | 5% | >40 |
The phenotypic APC ratio reflects the severity of the hypercoagulable state and provides information on the thrombotic risk associated with inherited and possibly acquired APC resistance. A modified APC resistance test, Chromogenix Coatest® APC™ Resistance V, is available which exclusively detects factor V-related APC resistance, i.e. APC resistance due to the FV:Q506 mutation. The assay modification involves a predilution of plasma samples with an excess of stabilized factor V-deficient plasma (V-DEF Plasma) containing a heparin antagonist. Since the predilution with VDEF Plasma normalizes the basal APTT reaction, it safely allows for APC resistance-testing of plasma from patients on oral anticoagulant or heparin therapy. It also produces a complete discrimination for FV:Q506, which makes the modified assay highly suitable for factor V mutation screening. The two APTT-reactions obtained from the modified APC resistance assay are expressed as an APC-V ratio, calculated in the same way as the APC ratio obtained from the classic test. The APC-V ratio provides genotypic information concerning factor V and is generally lower than the APC ratio for the same sample, regardless of the instrument used. Typical APC-V ratio ranges for different factor V genotypes are 2.2 – 3.2 for normal FV:R506, 1.4 – 1.8 for heterozygous FV:Q506, and 1.1 – 1.3 for homozygous FV:Q506.
Distribution of the FV:Q506 mutation in the world population
World distribution of APC resistance and FV:Q506
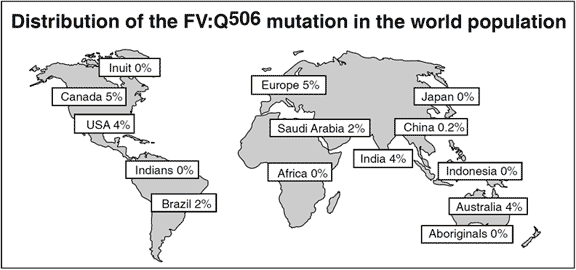
An overall cumulative analysis of different patient groups with venous thromboembolism shows that the prevalence of APC resistance is about 20% (range 0-64%). The variation in the prevalence of APC resistance between clinical studies are related mainly to differences in selection criteria and the uneven distribution of the FV:Q506 allele in the general population in different parts of the world. The highest prevalences of APC resistance and the FV:Q506 mutation have been found among healthy controls in several European populations of Caucasian origin, most notably in Cypriot Greek (13%), Swedish (11%), French (10%), British (9%), German (9%) and Dutch (5%)10 people. In contrast, the mutation appears to be rare among Chinese and absent among Japanese and Africans (Negroid). This could account for the relatively low incidence of venous thromboembolism reported these ethnic populations. The average FV:Q506 carrier frequency among healthy European controls is about 5%. Approximately 0.1% of a Caucasian population can be expected to be homozygous for this mutation.
FV:Q506 mutation determined using a DNA-based assay
| Country / region | FV:Q506 Ref. | n/n test. | % |
|---|---|---|---|
| Australia, Aboriginal | 0/73 | 0 | 77 |
| Brazil, Indians | 0/83 | 0 | 81 |
| Brazil, Blacks | 1/137 | 0.7 | 81 |
| Brazil | 2/100 | 2 | 57 |
| Canada | 19/356 | 5.3 | 82 |
| China, Han | 1/618 | 0.2 | 83 |
| China, Hong Kong | 0/293 | 0 | 70 |
| Finland | 4/137 | 2.9 | 93 |
| France, Paris | 5/229 | 2.2 | 74 |
| France, Strasbourg | 17/176 | 9.7 | 75 |
| Germany, South | 14/180 | 7.8 | 85 |
| Germany, North-East | 58/814 | 7.1 | 86 |
| Germany, North-West | 18/190 | 9.5 | 72 |
| Greece | 17/203 | 8.4 | 80 |
| Greek Cypriots | 25/187 | 13 | 77 |
| Greenland, Inuit | 0/133 | 0 | 87 |
| Iceland | 3/96 | 3.1 | 77 |
| India, North | 3/70 | 4.3 | 88 |
| Indonesia, Sumatra | 0/105 | 0 | 77 |
| Italy | 9/344 | 2.6 | 89 |
| Japan | 0/192 | 0 | 76 |
| Jamaica | 0/91 | 0 | 77 |
| Kenya | 0/60 | 0 | 77 |
| Mongolians | 0/36 | 0 | 77 |
| Netherlands | 14/474 | 3.0 | 54 |
| Papua New Guinea | 0/95 | 0 | 77 |
| Peru, Indians | 0/19 | 0 | 77 |
| Saudi Arabia | 5/200 | 2.5 | 90 |
| Senegal | 0/96 | 0 | 77 |
| Sweden | 11/101 | 11 | 78 |
| Taiwan, Aboriginals | 0/83 | 0 | 77 |
| United Kingdom | 21/237 | 8.9 | 77 |
| United Kingdom | 5/144 | 3.5 | 73 |
| USA, Blacks | 3/214 | 1.4 | 91 |
| USA | 42/704 | 6.0 | 69 |
| Zambia | 0/95 | 0 | 77 |
APC resistance phenotype determined using the classic APC resistance test
| Country | APC resistant n/n test. | % | ref. |
|---|---|---|---|
| Austria | 1/50 | 2 | 55 |
| France | 1/75 | 1.3 | 12 |
| France | 2/50 | 4 | 56 |
| Italy | 20/1212 | 1.2 | 64* |
| Japan | 3/291 | 1.0 | 59 |
| Netherlands | 14/301 | 4.6 | 10 |
| Poland | 1/110 | 0.9 | 62* |
| Spain | 3/107 | 2.8 | 65 |
| Sweden | 9/130 | 6.9 | 11 |
| USA | 2/39 | 5.1 | 67 |
APC resistance due to FV:Q506 in USA determined using the modified APC resistance test
| USA ethnic origin | US pop. millions | % FV:Q506 mutation |
|---|---|---|
| Caucasians | 185 | 5.3 |
| Hispanic | 25.8 | 2.2 |
| Afroamericans | 32.7 | 1.4 |
| Native Indians | 2.1 | 1.3 |
| Asians | 7.5 | 0.46 |
The origin of the FV:Q506 mutation
Several investigators have suggested that the high prevalence of the FV:Q506 mutation could be due to the evolutionary advantage it would confer, which has helped to maintain and spread the mutation. It is possible that the selective disadvantage of a life-long hypercoagulable state could be balanced by, for example, the protection against excessive blood loss during delivery and menstruation. The selective risk of the FV:Q506 mutation would also be of less historical importance as people in ancient times were not exposed to modern risk factors for thrombosis (e.g. oral contraceptives, surgery, sedentary lifestyle etc.). The high allelic frequency of FV:Q506 in Caucasian populations and its linkage to different polymorphisms in the factor V gene, supports the hypothesis that the mutation occurred as a single event in the ancient European population. The time of this event would be approximately 30,000 years ago, i.e. after the diversion of Africans from non-Africans (140,000 years ago) and after the diversion of Caucasians from Mongolic populations (70,000 years ago), but before the diversion of Caucasian subpopulations. However, since the FV:Q506 mutation involves a CpG dinucleotide, which is an established hotspot for mutation, the possibility of recurrent mutations in other races should not be ruled out altogether.
Clinical manifestations of APC resistance
The clinical manifestations of inherited, heterozygous protein defects in familial thrombophilia involving antithrombin, protein C, protein S and factor V (APC resistance) are fairly similar. Mutations affecting the qualitative or quantitative function of these proteins often result in venous thromboembolism at a young age (before the age of 45 years) and are followed by a tendency towards recurrent thrombotic episodes. The most common manifestation of APC resistance is deep venous thrombosis (DVT) of the lower limbs, with or without pulmonary embolism, which accounts for about 90% of all thrombotic episodes. Other, less frequent, manifestations include superficial thrombophlebitis and unusual sites for thrombosis such as the mesenteric, central retinal, portal, internal jugular, and cerebral veins.
Thrombotic risk
The relative risk of DVT for carriers of the FV:Q506 mutation compared to non-carriers has been estimated to increase eight-fold for heterozygotes (single defect) and 80-fold for homozygotes (double defect). Since aging itself is a risk factor for thrombosis, the absolute risk increases with age. A similarly increased risk of pulmonary embolism has been observed by some investigators, although this could not be confirmed by others. The risk of recurrent venous thromboembolism in carriers of the FV:Q506 mutation has been reported to be three to fivefold higher compared to patients without the mutation, but again there is no consensus on this matter.
FV:Q506 is a mild risk factor by itself
The penetration of clinical manifestations among APC resistant individuals is variable, and a majority of heterozygous carriers of FV:Q506 actually never experience any symptoms. In fact, not even homozygous carriers will necessarily be affected by thrombosis during their lifetime. These facts illustrate that FV:Q506 is a mild risk factor per se and that the probability of APC-resistant individuals developing thrombosis is dependent on the coexistence of other risk factors. About 60% of APC-resistant patients have their first thrombotic event in combination with pregnancy, oral contraceptives, trauma or surgery. Furthermore, because of the high prevalence of APC resistance in the general population, its combination with other genetic defects is not unusual. A wide range of disorders have been reported in connection with APC resistance, implicating its part in the development of thrombotic complications. These include the Budd-Chiari syndrome, nephrotic syndrome, leg ulcers, heparin-induced thrombocytopenia, priapism, polycythemia vera, essential thrombocythemia, child-thrombosis, cutaneous skin necrosis, neonatal purpura fulminans, acute lymphoblastic leukemia, systemic sclerosis, and preeclampsia.
Prevalence of APC resistance phenotype in patients with venous thrombosis
| Country | Venous thrombosis n pts. | Venous thrombosis n APC res. | References |
|---|---|---|---|
| Austria | 40 | 7 (17%) | Halbmayer et al |
| France | 175 | 29 (17%) | Trossaërt et al |
| France | 48 | 9 (19%) | Cadroy et al |
| France | 183 | 24 (13%) | Samaha et al |
| Italy | 20 | 2 (10%) | *Tosetto et al |
| Italy | 118 | 33 (28%) | De Stefano et al |
| India | 28 | 6 (21%) | Pati et al |
| Japan | 43 | 5 (12%) | Kambayashi et al |
| Japan | 22 | 4 (18%) | Fujimura et al |
| Netherlands | 301 | 64 (21%) | Koster et al |
| Poland | 72 | 9 (12%) | *Lopaciuk et al |
| Sweden | 104 | 34 (33%) | Svensson et al |
| Spain | 72 | 3 (4%) | *Borell et al |
| Spain | 176 | 14 (8%) | Ortega et al |
| USA | 25 | 16 (64%) | Griffin et al |
| USA | 37 | 9 (24%) | Chusman et al |
| Total | 1,464 | 268 (18%) |
Prevalence of the FV:Q506 mutation in patients with venous thrombosis according to gene analysis
| Country | Venous thrombosis n pts. | Venous thrombosis n FV:Q506 | References |
|---|---|---|---|
| USA | 121 | 14 (11%) | Ridker et al |
| Japan | 22 | 0 (0%) | Fujimura et al |
| France | 87 | 14 (16%) | Alhenc-Gelas et al |
| Netherlands | 301 | 53 (17%) | Bertina et al |
| Netherlands | 27 | 10 (37%) | Voorberg et al |
| Netherlands | 471 | 92 (19%) | Rosendaal et al |
| Brazil | 40 | 8 (20%) | Arruda et al |
| Australia | 45 | 12 (26%) | Ma et al |
| Total | 1,114 | 203 (18%) |
Prevalence of the FV:Q506 mutation in patients with arterial thrombosis compared with controls according to factor V gene analysis
| Country | Arterial thrombosis n pts. | Arterial thrombosis n FV:Q506 | Controls | References |
|---|---|---|---|---|
| Australia | 222 | 11 (5.0%) | (4%) | van Bockxmeer et al |
| Finland | 358 | 16 (4.5%) | (2.9%) | Kontula et al |
| Germany | 224 | 21 (9.4%) | (4.1%) | März et al |
| Sweden | 101 | 18 (18%) | (11%) | Holm et al |
| UK | 386 | 16 (4.1%) | (5.6%) | Catto et al |
| USA | 583 | 32 (5.5%) | (5.5%) | Ridker et al |
Arterial thromboembolism
Although there is a clear link between APC resistance due to the FV:Q506 mutation and venous thrombosis, the same link to arterial thrombosis is enigmatic. Several studies have reported the presence of APC resistance in young stroke patients, suggesting that it contributes to the pathophysiology. Halbmayer et al for example, found that 20% (6 out of 30) of young Austrian stroke patients were APC-resistant according to the classic APC resistance test. In contrast, other studies of stroke patients showed no increased prevalence of either the APC resistance phenotype or FV:Q506 mutation compared to healthy controls.
The possible correlation between the FV:Q506 mutation and the risk of ischemic heart disease, particularly myocardial infarction, has also been investigated. With the exception of two papers, the general conclusion is that the FV:Q506 mutation is not an important risk factor for arterial thrombosis in heterozygotes, but may have a role in homozygotes. Some papers have appeared recently which may help to clarify the situation. These suggest that acquired (or inherited) APC resistance, independent of the FV:Q506 mutation, may indeed be an important risk factor for arterial thrombosis. These observations are most interesting and they call for an evaluation in different patient groups, using the classic APC resistance test, to explore whether the APC ratio may be predictive for both venous and arterial thrombotic events.
APC ratios in thrombophilic families
Zöller et al investigated 50 thrombosis-prone Swedish families with APC resistance. In three of these families the FV:Q506 mutation was not present, suggesting another, as yet unidentified, cause of APC resistance. In total, 308 family members were investigated; 146 normal, 144 heterozygotes and 18 homozygotes. APC ratios were low in all the homozygous and most of the heterozygous cases. APC ratios in the APC-resistant individuals who lacked the mutation ranged from 1.3 to 2.0. Heterozygotes with a history of thrombosis had significantly lower APC ratios than those without thrombosis and none of the heterozygotes with APC ratios >2.0 had experienced thrombosis. Moreover, relatives without the mutation but with thrombotic histories had on average lower APC ratios than those without thrombosis. Significant differences in thrombosis-free survival curves and APC ratios were observed between the groups, thus confirming that APC resistance is an important risk factor for thrombosis. By the age of 33, 8% of the normals, 20% of the heterozygotes and 40% of homozygotes had experienced manifestations of venous thrombosis. The average age for the first thrombotic event was 25 (range 10 to 40 years) for homozygotes and 36 (range 18 to 71 years) for heterozygotes. In the thrombosis-prone families the observed incidence of thrombosis was higher than expected, suggesting that these families have been affected by additional genetic defects.
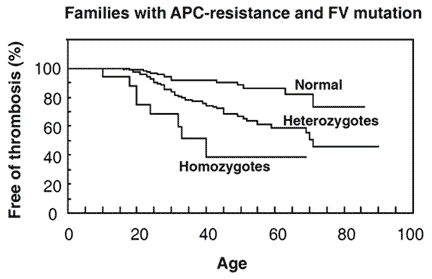
genotypes. The probability of being free from thrombotic events at a certain age in family members without the FV:Q506 mutation, compared with family members with the mutation. At the age of 33, 8% of normals, 20% of heterozygotes and 40% of homozygotes had venous thrombotic events. Reproduced by permission of Zöller et al and The American Society for Clinical Investigation.
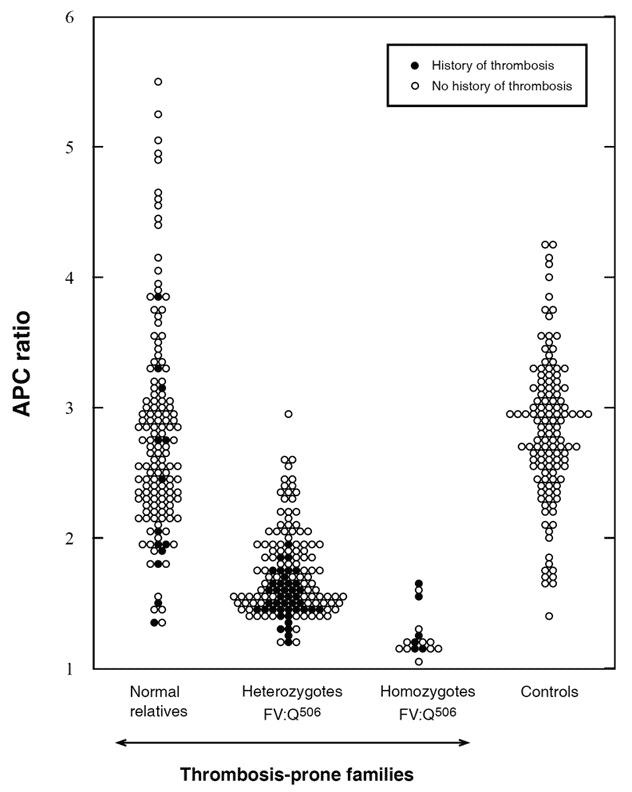
APC ratios in non-anticoagulated carriers of the FV:Q506 mutation and in family members without the mutation (normals), compared with unrelated healthy controls. The APC ratios were determined by the original APC resistance test method. Using a cut-off value of 2.0, the sensitivity and specificity for the FV:Q506 allele would be 85% and 87%, respectively. APCratios (mean ±SD) in normals 2.8 ±0.8, heterozygotes 1.7 ±0.3, homozygotes 1.3 ±0.2, and controls 2.8 ±0.6. Reproduced by permission of Zöller et al and The American Society for Clinical Investigation.
Multiple genetic defects
The underlying cause of familial thrombophilia has long been considered to be single-gene defects. However, the notion that this idea was too simple has been reported repeatedly in recent years, particularly in connection with protein C deficiency. It was found that the same type of mutation could affect different families differently, giving rise to the idea that several genetic risk factors in combination are usually needed for clinical manifestations to occur. Strong evidence supporting this view came with the discovery of APC resistance. Its high prevalence in the general population and the observation that individuals with combinations of inherited risk factors (e.g. FV:Q506 and protein C deficiency) suffer more severely from thrombosis, and at a younger age, than those with single defects, has led to the idea of familial thrombophilia being primarily a polygenetic syndrome. Most studies have confirmed this by showing a relatively high incidence of FV:Q506 among clinically symptomatic probands in thrombophilic families with protein C, protein S, or antithrombin deficiency. The observed frequency variation of the FV:Q506 mutation among these probands is probably related to population differences. Other interesting candidates for polygenetic familial thrombophilia involving FV:Q506, include hyperhomocysteinemia (due to either cystathione-b-synthase or methylenetetrahydrofolate reductase deficiency), familial antiphospholipid syndrome, heparin cofactor II deficiency, plasminogen deficiency, and possibly also elevated prothrombin levels due to a 20210 AG genotype in the prothrombin gene.
The incidence of thrombotic episodes in thrombophilic families, related to the number of genetic defects (‘hits’)
| One hit | One hit | Two hits | Ref |
|---|---|---|---|
| 20% FV:Q506 | 54% AT def. | 92% FV:Q506 + AT def. | 150 |
| 13% FV:Q506 | 31% PC def. | 73% FV:Q506 + PC def. | 147 |
| 19% FV:Q506 | 19% PS def. | 72% FV:Q506 + PS def. | 148 |
Influence of FV:Q506 in hereditary bleeding disorders
The influence of FV:Q506 on inherited bleeding disorders has also been studied. A possible moderation of the hemophilia A (factor VIII deficiency) phenotype has been observed in some cases, although this was not seen in others. A more surprising influence of FV:Q506 is seen in cases of parahemophilia (factor V deficiency). Heterozygotes for this rare bleeding disorder are often asymptomatic, since they have one functional factor V allele that maintains adequate factor V levels in blood (about 50%). However, the coinheritance of a factor V deficiency mutation on one allele and FV:Q506 on the other leads to a severe APC resistance phenotype similar to the homozygous FV:Q506 state.
APC resistance and circumstantial risk factors for thrombosis
Pregnancy
During normal pregnancy the plasma concentrations of several of the proteins involved in the hemostatic mechanism change towards a hypercoagulable state. Although these changes are of physiologic importance in minimizing the risk of blood loss at delivery, they also increase the risk of thrombotic complications. In developed countries the overall incidence of thrombosis has been reported to be around 0.09% during pregnancy, with the risk being two to three-fold increased during puerperium. APC resistance appears to be an important predisposing risk factor for thrombosis in connection with pregnancy. In two Swedish studies, 45-60% of women with a history of pregnancy-related thrombosis were found to be APC resistant. Carriers of the FV:Q506 mutation appeared to be especially prone to developing thrombosis in early pregnancy and after delivery, compared to non-carriers of the mutation. APC resistance also seems to be associated with an increased risk of second trimester miscarriage related to placental infarction, and may be involved in the mechanism of preeclampsia.
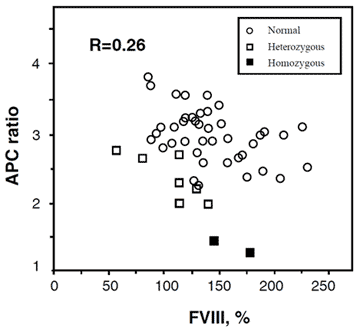
Elevated factor VIII levels
In general, women have slightly lower APC ratios compared to men. This difference becomes more pronounced during pregnancy and indeed a substantial proportion of pregnant women may even develop an acquired APC resistance. Although it has been demonstrated that increased factor VIII levels lower the APC ratio, and that increased factor VIII levels are common during pregnancy, inflammatory diseases, and the use of oral contraceptives, other hormonally-influenced factors probably contribute since the actual correlation between the APC ratio and the concentration of factor VIII is low.
Until more is known about the mechanism and clinical relevance of acquired APC resistance, the influence of pregnancy and sex-hormones should be taken into consideration when interpreting the APC ratio obtained using the classic APC resistance test. The detection of factor V related APC resistance during pregnancy using the modified APC resistance test is straightforward, since predilution in factor V-deficient plasma normalizes any pregnancy-induced changes in the patient’s plasma.
Oral contraceptives
The use of oral contraceptives (OCs) is a much-debated risk factor for thrombosis. Numerous studies have shown that oral contraceptives increase the risk of thrombosis about two to nine-fold depending on the active substance used, compared to non-users. However, it is still largely unclear why oral contraceptives cause thrombosis in some women. Similar to pregnancy, increased levels of procoagulant factors, as well as reduced APC ratios and reduced levels of antithrombin and free protein S have been reported in women using OCs. For the majority these procoagulant changes are negligible, although for a small number of women with a genetic or acquired predisposition for thrombosis, the added prothrombotic influence of OCs may be sufficient to trigger a thrombotic event.
The risk of venous thrombosis in OC users with APC resistance has been investigated by Vandenbroucke et al. The risk of thrombosis among OC users in this study was shown to increase 4-fold when compared to women not using OCs (baseline risk about 0.01% annually). Women heterozygous for the FV:Q506 mutation who did not use OCs showed an 8-fold increase in thrombotic risk, whereas a 35-fold increase in risk was shown in OC users heterozygous for the mutation. Thus, the joint effect of the two risk factors appears to be multiplicative. Through a similar effect, the increase in risk for homozygotes using OCs is several 100-fold. Clinical studies also demonstrate that OCs leads to an unacceptably high risk of venous thrombosis in females with homozygous FV:Q506.
Relative risks associated with oral contraceptives (OCs) and the factor V:Q506 mutation
| Genotype | Non-user – | OCs (overall) | OCs (levonorgestrel) | OCs (desogestrel) |
|---|---|---|---|---|
| Normal | 1 | 4 | 4 | 9 |
| Heterozygotes | 8 | 32 | 15 | 48 |
| Homozygotes | 80 | 320 | 150 | 480 |
Hellgren et al investigated 28 women with a history of thrombosis in connection with OCs and they found that nine (32%) of the women had APC ratios <2.0, indicating APC resistance. None of the women investigated were using OCs at the time the blood samples were taken and in only one of nine patients with APC resistance was an additional risk factor for thrombosis identified.
Considering the high prevalence of APC resistance in the general population, a substantial number of women are placed at a higher risk by using OCs. In North America alone there are about 10 million OC users. Assuming the allele frequency for FV:Q506 is 0.025, then roughly 500,000 would be heterozygous and 6,000 homozygous for the mutation. Since the annual thrombotic risk is about 0.3% for heterozygotes and 3% for homozygotes using OCs, it can be calculated that around 2,000 thrombosis cases per year in North America are caused by the combination of APC resistance and OCs. This figure should be compared to the number of thrombosis cases in OC users without the mutation, which is about 4,000 cases per year. Thus, in the presence of both risk factors (OCs + mutation), venous thrombosis appears to develop in a substantial number of women who would never have had thrombosis in the presence of either risk factor alone. It can be estimated that 1-2% of all cases of venous thrombosis caused by OCs will have a fatal outcome due to pulmonary embolism.
Physicians who prescribe OCs generally interview the woman about her family history to learn whether there are any cases of thrombosis, indicating a genetic predisposition. However, since many homozygous APC resistance patients have asymptomatic parents that are heterozygous, it would not be suspected from interviewdata alone. This fact raises the question as to whether all women should be screened for the FV:Q506 mutation prior to prescribing OCs. The potential reduction in mortality and morbidity cases due to thromboembolism caused by the pill must, however, be weighed against the risk of thrombosis in connection with unwanted pregnancy.
Surgery
Patients undergoing major trauma or surgery, in particular orthopedic or abdominal surgery, generally run a high risk of experiencing thromboembolic events due to a sustained activation of coagulation. The precise risk for an individual is determined not only by the type, extent and duration of the surgical trauma, but also by the accumulation of predisposing risk factors such as advanced age, morbidity and thrombophilia. The general and standardized use of heparin for post-operative thromboprophylaxis has reduced thrombotic complications to a great extent during recent decades. However, it still occurs in a substantial number of patients. Thus, it may be beneficial to pre-operatively identify these individuals at higher risk in order to adopt a more individualized and adequate prophylaxis. The latter approach is supported by recent studies, showing that prolonged prophylaxis using LMW heparin in connection with hip replacement resulted in a 50% reduction in venographically verified deep venous thrombosis. More studies are needed in this area to evaluate whether individualized prophylactic treatment might be beneficial in patients with genetic risk factors for thrombosis.
Risk groups in trauma and surgery in order of decreasing frequency of DVT using no thrombo-prophylaxis
| Spinal Cord Injury | 70-75% |
| Knee arthroplasty | | |
| Leg amputation | | |
| Hip fracture surgery | | |
| Hip arthroplasty | | |
| Lower limb fracture | | |
| Open prostatectomy | | |
| General abdominal surgery | | |
| Gynecological surgery | | |
| Kidney transplantation | | |
| Noncardiac thoracic surgery | ↓ |
| Neurosurgery Open meniscetomy | 20-25% |
At present, the underlying role of APC resistance in postoperative thrombosis is under intense research. In a Danish study it was found that 30% of patients who developed deep venous thrombosis after knee arthroplasty were APC resistant. APC resistance was also shown to be a risk factor for arterial reocclusion of vascular grafts in peripheral vascular surgery. These studies are highly interesting and suggest that it would be cost-benefical to screen for APC resistance pre-operatively in order to reduce post-operative thrombosis.
Antiphospholipid antibodies
Lupus anticoagulant and anticardiolipin antibodies are acquired antiphosholipid antibodies (APAs) which were initially described in patients with systemic lupus erythematosus (SLE). Recently, the interest in these types of antibodies, which are more correctly described as antibodies directed against phospholipid-protein complexes, has focused mainly on their strong correlation with thrombotic disease, thrombocytopenia and recurrent fetal loss. The association of one of these complications and the presence of APAs has been termed the antiphospholipid syndrome. The mechanism underlying the syndrome is at present unknown, although various interferences in the protein C pathway have been suggested as being a possible pathogenic mechanism for thrombosis One such interference may be the selective blocking of APC on phospholipid surfaces by specific types of APAs, leading to an acquired APC resistance. Several reports have also demonstrated acquired APC resistance to be a relatively common feature in patients with the antiphospholipid syndrome, even when the uncertainties of lowered APC ratios caused by APA-induced prolongation of the basal APTT were considered. At present, there is no clear clinical evidence to support a higher thrombotic risk among patients with the antiphospholipid syndrome and acquired APC resistance. Furthermore, the FV:Q506 mutation does not seem to be over-represented in patients with the syndrome compared with healthy controls.
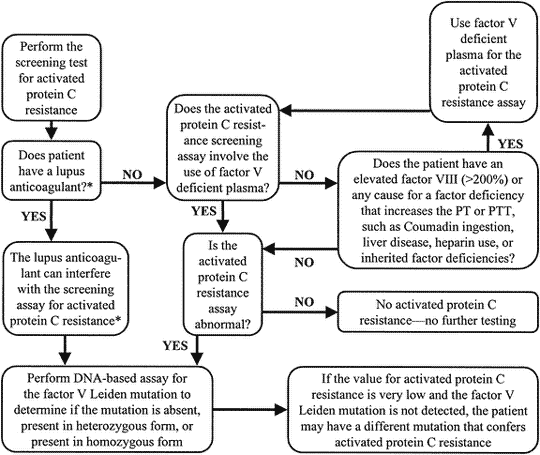
When to test for APC resistance phenotype and FV:Q506 genotype
Up to now, the classic APC resistance test has been used mainly as a simple screening assay for the FV:Q506 mutation. However, several limitations have been reported for this application. Firstly, this test has a sensitivity and specificity for the mutation which is usually in the range 75-90%. Secondly, the test is only reliable if the basal APTT-reaction is within the normal range, which therefore disqualifies many APC-resistant patients on anticoagulant therapy from testing.
As the classic APC resistance test stands today, it is not recommended for FV:Q506 mutation screening. The classic test provides phenotypic information about APC resistance and should primarily be used as a complimentary test to the modified APC resistance test in a thrombosis investigation. A preliminary guideline for how to use the classic and modified APC resistance test is given below.
Preliminary guideline for APC resistance and/or FV:Q506 testing
- Caucasian patients with venous thrombosis should be tested, since the risk in APC resistant individuals is life-long. The identification of a defect is useful in decisions concerning anticoagulant therapy.
- First-degree relatives to thrombosis patients with inherited APC resistance should be tested as this may help in estimating the thrombotic risk profile of the family and the prevention of future thrombotic events.
- Thrombosis patients known to carry other genetic defects associated with thrombophilia should be tested for APC resistance, as symptomatic individuals tend to have more than one defect.
- FV:Q506 screening of patients with arterial thrombosis is not warranted except for young patients.
- General screening for FV:Q506 may prove to be beneficial before exposure to circumstantial risk factors such as prior to the prescription of oral contraceptives.
APC resistance phenotyping
Interestingly, the APC ratio is not a simple, one-variable reflection of the APC response in vivo. Instead, it seems to reflect an anticoagulant system response that may decrease under a hypercoagulable state. A poor anticoagulant response to APC, independent of the FV:Q506 mutation, could therefore be a thrombotic risk factor or risk marker in a wide range of conditions and circumstances including venous thromboembolism, antiphospholipid protein syndrome, second trimester miscarriage, systemic sclerosis, ischemic stroke, occlusion after vascular surgery, pregnancy and the use of oral contraceptives. Future studies will clarify whether the phenotypic APC ratio obtained from the classic test may serve as a predictor of venous and arterial thrombotic events.
Screening for FV:Q506
The modification of the APC resistance test, in which sample plasma is prediluted in an excess of stabilized factor V-deficient plasma, improves the discrimination for the FV:Q506 mutation dramatically. The modification also allows for the testing of patients on anticoagulant therapy and strongly reduces the influence of preanalytical variables such as storage and plasma handling. Evaluation of this test using different categories of clinical samples showed that the specificity and sensitivity for the presence of the FV:Q506 mutation was 100%. The robustness and simple format of the modified test, together with its high discrimination between normal and mutated factor V genotypes, makes it an ideal tool for FV:Q506 mutation screening. An important point when prediluting the sample plasma in factor V-deficient plasma is that phenotypic expressions related to factors other than factor V are lost. In order to obtain phenotypic and genotypic information for a correct thrombophilia diagnosis, one approach could be to analyze each plasma sample using both the classic and the modified APC resistance test. This strategy identifies APC-resistant patients, both with and without the FV:Q506 mutation, and keeps the need for confirmatory gene analysis to a minimum.
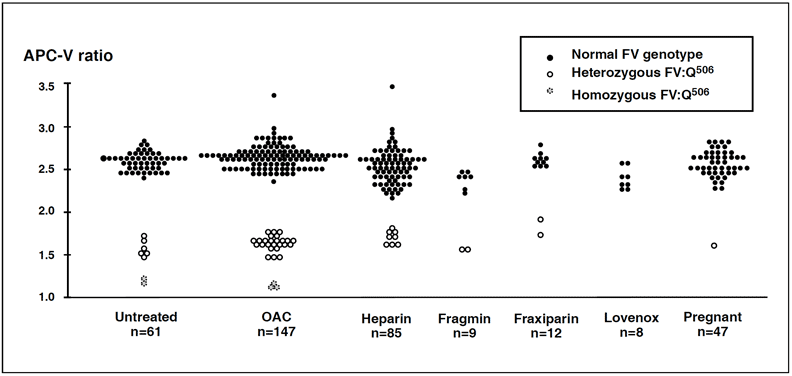
Management of APC resistant patients
A potentially large number of thrombosis-prone individuals with APC resistance will most likely be identified in the near future. This of course raises the question of patient management. At present there are no established guidelines for managing thrombotic patients with APC resistance, although it is generally agreed that they should be treated in the same way as the patients with antithrombin, protein S and protein C deficiency. An acute thrombotic episode should be treated conventionally with heparin for 5 to 10 days, followed by an oral anticoagulant (warfarin) within 24 hours to produce an International Normalized Ratio (INR) of 2.0 to 3.0. Patients should be given general advice on how to minimize the thrombotic risk, including dietary advice, cessation of smoking and avoiding long periods of immobility. Thrombophilic women should avoid oral contraceptives and all patients should be notified that they may require special treatment prior to surgical, medical or obstetric procedures that carry an increased thrombotic risk.
Anticoagulant treatments require individual considerations
Because only a proportion of the subjects with a heterozygous defect develop thrombosis, it is unjustifiable to put symptom-free APC resistant individuals on thromboprophylaxis solely on the basis of having a genetic defect. It is, however, essential that asymptomatic individuals in thrombosis-prone families are carefully counseled with respect to their defect and offered short-term prophylaxis in special situations where there is an extra risk of thrombosis. Women with a history of thrombosis and who have a known genetic defect may require anticoagulation throughout pregnancy, preferably by using dose-adjusted subcutaneous heparin. As a rule, all thrombophilic women should be offered thromboprophylaxis in conjunction with delivery and puerperium. Homozygous and heterozygous patients with a second anticoagulant defect should be given preventive therapy in all risk situations and long-term therapy should be considered if thrombosis is recurrent.
The prescription of oral contraceptives
When a woman has experienced venous thrombosis after oral contraceptive use, it is recommended that she is tested for the possible presence of the FV:Q506 mutation. If she is heterozygous for the mutation she should be carefully informed about her thrombotic risk and counseled about the type of contraceptive she should use in the future. The mere fact that she has had thrombosis indicates that the risk in her case is significant. If she is homozygous for the mutation, she should be strongly recommended to discontinue the use of oral contraceptives. As in any investigation of a young patient with venous thrombosis and APC resistance, it is also recommended to search for other causes of inherited thrombophilia.
Summary
The APC resistance phenotype is mainly diagnosed using the classic APTT-based test, which detects both inherited and acquired APC resistance. A modified APTT-based test using predilution of sample plasma with factor V deficiency plasma has been developed with high discrimination for factor V-related APC resistance. The factor V:Q506 mutation is highly prevalent among Caucasians (2-13%), it is present among people of Northern India (4%) and Saudi Arabia (2%), but is very rare among, for example Japanese, Eskimoes and Australian aborigines. The race-dependent distribution of the mutation may explain the higher incidence of thrombosis reported for Caucasians. The most common clinical manifestation of APC resistance is deep venous thrombosis, with or without pulmonary embolism. The relative risk of venous thrombosis for carriers of the factor V:Q506 mutation has been estimated to increase 8-fold for heterozygotes and 80-fold for homozygotes. The mutation does not seem to be an important risk factor for arterial thromboembolism, although acquired (or inherited) APC resistance not attributable to the mutation may be a risk factor for stroke as well as for venous thrombosis. The current recommendations for treating APC resistant patients are similar to those for other established inherited thrombophilic defects.
APC Resistance Assay Methods
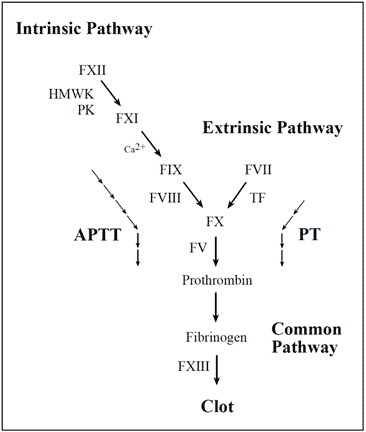
Laboratory determination of APC resistance
The laboratory analysis of poor anticoagulant response to APC is based on an activated partial thromboplastin time (APTT) assay, which is modified in the manner described by Dahlbäck et al. The standard APTT reaction begins by adding a surface-activating agent (e.g. kaolin, silica, ellagic acid) and a phospholipid preparation to citrated, platelet-poor plasma, thereby achieving maximum activation of factor XI. The plasma is then recalcified in order to activate the coagulation cascade and the time for clot formation is measured. The name APTT originates from the fact that the phospholipid reagents were originally derived from a lipid-enriched extract of complete thromboplastin (now called tissue factor), hence the term partial thromboplastin. The APTT reaction is dependent on factors of both the intrinsic and the common pathway of the
coagulation system.
The classic APC resistance test
In the classic APC resistance test (available as Chromogenix Coatest® APC™ Resistance), two APTT reactions are performed, one in the presence of a carefully defined amount of APC and the other in its absence. The result can be calculated either as a prolongation of the clotting time in presence of APC or as the ratio between the clotting times in the presence and absence of APC. Use of the APC ratio for expression of the results is preferred, mainly due to its lesser susceptibility to preanalytical sample handling variations.
As for any APTT-based assay, it is important to follow a standardized procedure for blood sampling and storage. The APTT reaction without the addition of APC should be within the normal range (25-40 sec) in order to obtain valid APC ratios. This means that plasma from patients undergoing therapies causing deficiencies in clotting factors, for example treatment with warfarin or heparin, will not allow for reliable APC resistance analysis. Anticoagulant therapies must therefore, if possible, be discontinued for at least one week to allow re-establishment of the baseline prothrombin level. An alternative method in the case of heparin therapy is to neutralize heparin in the sample using a heparin antagonist.
Instrument effects
The APC ratios obtained from the analysis, using the Chromogenix Coatest® APC™ Resistance kit, of plasmas from healthy individuals on different coagulation instruments is typically within the range 2-5. The precise cut-off value for a diagnosis may vary slightly due to the instrumentation and the condition of the specific instrument. In general, coagulation instruments with a turbidimetric or photometric clot detection principle offer better discrimination than instruments using an electromechanical detection principle.
The normal APC ratio range
Since the cut-off ratio for an abnormal APC response may vary slightly between laboratories and instruments, it is recommended that laboratories establish their own normal range. The normal range can be determined by measuring the APC ratios from at least 50 healthy non-APC resistant individuals in the age range 20-65 years and calculating the median APC ratio. The APC ratio cut-off is then obtained by multiplying the median value by either 0.75 (median above 3) or by 0.8 (median below 3). Alternatively, the cut-off can be calculated as the mean minus two standard deviations. If it is not possible to perform factor V genotyping in the determination of the 50 healthy non-APC resistant individuals, a practical approach is to exclude the lowest 10% and highest 10% of the APC ratios before calculating the APC cut-off ratio.
Normalized APC ratio
In addition to the instrument effect on the APC ratio, different sources of APTT reagents as well as different batches of the same APTT reagent may show varying sensitivity to APC. In order to compare results from different laboratories accurately it may be advantageous to present APC ratios after having normalized them against a pooled normal plasma, PNP (APC ratio/PNPAPC ratio). The properties of the PNP are important to consider as mixing studies of different plasmas show that a 10% contribution or more of APC-resistant plasma in the PNP greatly affects the normalized APC ratios obtained.
Effect of plasma preparation and handling
To ensure reliable results it is necessary to obtain proper control of preanalytical variables. Thus, blood samples must be centrifuged thoroughly and the plasma removed carefully in order to minimize the number of platelets in the sample. The main reason for this being that freeze-thaw cycles reduce the APC ratios in the presence of platelets due to rupture of platelet membranes leading to the exposure of procoagulant phospholipids and platelet- bound factor V. As a rule, samples should be handled in the same way as the controls used to establish the normal range, i.e. ”like should be compared to like”.
A routine single centrifugation of the blood sample at 2,000 x g for 20 minutes will normally be adequate to obtain platelet-poor plasma (<1% normal platelet count). Centrifugation should take place at room temperature in order to avoid cold activation since this may eventually lead to the activation of factors VIII and V. Careful separation of plasma from platelets is achieved by leaving 0.5-1.0 cm of plasma above the cell layer. If plasma is to be frozen, it should be frozen rapidly in volumes of £ 1ml, at -20 °C or below.
Antiphospholipid antibodies
The presence of acquired antiphospholipid antibodies, such as lupus anticoagulant, is a relatively common finding in thrombophilic plasma. Because the antibodies react with anionic phospholipids they may give rise to a prolonged clotting time and thus may influence the APC ratio (decreased ratio). It has been observed that the effect of lupus anticoagulants can be surmounted by the addition of excess phospholipid, however further studies are needed to confirm this observation.
Influence of factors V and VIII
Mixing experiments of different plasmas have demonstrated that changes in factor V levels between 12-100% do not have any significant effect on the APC ratio.
Very high factor VIII levels, for example in connection with pregnancy and inflammatory states, have been reported to lower the APC ratio, although the actual correlation between factor VIII activity and the APC ratio appears to be weak.
Influence of factors II, IX, X, protein S and protein C
Prothrombin and factor X concentrations below 50% tend to produce higher APC ratios. In contrast, the APC ratio is not influenced to any great extent by variations in factor IX and protein S concentrations down to 30% of normal. Variations in plasma levels of protein C have no influence on the APC ratio since a standardized amount of exogenous APC is added in the assay. On the other hand, it should be noted that the factor V:Q506 mutation may influence certain clotting-based functional protein S and protein C assays leading to the incorrect diagnosis of APC resistance as either protein S or protein C type II deficiency.
The modified APC resistance test
A modification of the classic APC resistance test has been described in which the sample plasma is mixed with an excess of factor V-deficient plasma in the range 1:5 to 1:20. A predilution stage normalizes the concentration of plasma proteins involved in the formation and regulation of thrombin, except for factor V, resulting in an improved discrimination for the factor V:Q506 mutation. Because prediluted plasma samples obtained from patients undergoing oral anticoagulant therapy have an APTT reaction within the normal range independent of treatment intensity, it is now possible to test for APC resistance due to factor V mutation in a large group of patients previously disqualified from APC resistance testing. The inclusion of a heparin antagonist such as Polybrene® in the factor V deficiency plasma also makes it possible to analyze samples with heparin levels £ 1 IU/ml (unfractionated and LMW preparations).
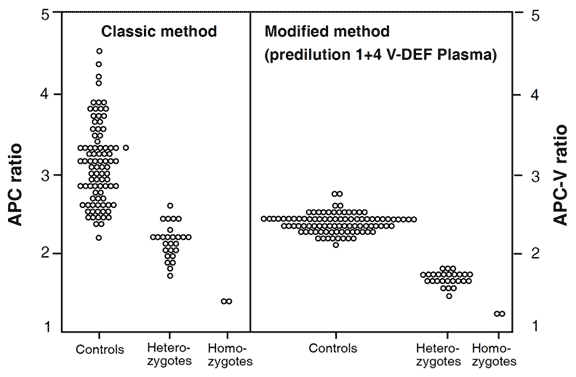
with APC-V ratios obtained from the modified test method. Both these methods
complement each other. The original method detects thrombosis-prone individuals
with APC resistance due to the FV:Q506 mutation, unknown mutations or acquired
factors and indicates the severity of their hypercoagulable state. The modified
method provides a high discrimination for the factor V:Q506 mutation and allows for
analysis of plasma from patients on oral anticoagulant or heparin therapy. The same
individuals were tested in both groups. Instrument ACL.
The use of a stabilized factor V-deficient prediluent (V-DEF Plasma) in the modified test, strongly reduces the influence of pre-analytical variations such as plasma handling and storage. Furthermore, no significant difference between fresh and frozen plasma is obtained and platelet counts up to 15,000/mL are easily tolerated. Predilution (1+4) reduces most sources of interference, although it cannot be excluded that the analysis of plasma from patients with antiphospholipid antibodies may result in an abnormal APTT. In such cases, increasing the dilution factor may correct the test result (e.g. 1+9 or 1+19). In neonates and infants (< 6 months of age) a predilution factor of 1+9 is needed because of their special hemostatic condition.
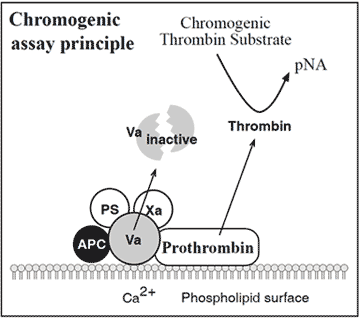
Alternative functional assays for APC resistance
The present detailed understanding of the mechanism behind APC resistance has produced a number of alternative assay concepts for APC resistance. A novel chromogenic APC resistance assay is presented below.
Different assay concepts for APC resistance
- APTT ± APC (± FV def. plasma)
- APTT ± Protac (± FV def. plasma)
- RVV ± APC
- RVV ± Protac
- FXa + APC
- Tissue factor/FV def.plasma ± APC
- Chromogenic using a FXa substrate
- Chromogenic using a thrombin substrate
Overview of functional methods for APC resistance. Explanations: ±APC = test with and without added APC, Protac = protein C activator, RVV = factor X activator, FV def =factor V deficient.
The assay is based on the addition of factor Xa to diluted test plasma in the presence of calcium ions, phospholipids and APC. Presence of the factor V:Q506 mutation decreases the rate of inactivation of factor Va by APC and allows for appreciable thrombin generation. This is measured by the hydrolysis of a chromogenic thrombin substrate. Preliminary results show complete concordance between the chromogenic assay and the modified APC resistance test.
PCR-based assays for FV:Q506
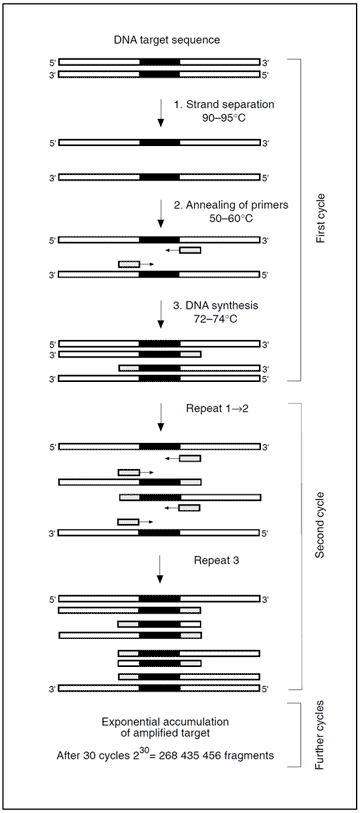
Identification of a mutation is generally accomplished by amplifying genomic material containing the mutation, followed by a mutation detection procedure. Amplification is achieved by the polymerase chain reaction (PCR) technique using either DNA or mRNA as a template. The key components in a standard PCR include a pair of oligonucleotide primers (around 20 bases long), the DNA building blocks; deoxyribonucleoside triphosphates, and a heat-resistant DNA polymerase (e.g. Taq). These components are added to a closed test tube containing the template DNA. A temperature-driven cyclic process is then initiated, consisting of three stages: 1) DNA strand separation, 2) Annealing of primers, and 3) DNA synthesis. A central point of the PCR is that all new DNA strands serve as templates in successive cycles. The new DNA, consisting of the target sequence flanked by primers, increases exponentially in subsequent cycles. After n cycles the DNA target sequence is amplified 2nfold. The amplification is a million-fold after 20 cycles and can be carried out in less than an hour.
In the seminal work by Bertina el al, in which the FV:Q506 mutation was first identified, the detection stage was performed by enzymatic digestion of the amplification product using the Mnl I restriction enzyme, followed by agarose gel electrophoresis. Presence of the mutation removes one of normally two cleavage sites for Mnl I. The resulting number and size of the fragments indicates whether the mutation is present or not.
Apart from methods involving direct sequencing of cDNA, a vast number of PCR-based methods for the determination of the factor V:Q506 mutation have been published. These involve improvements of the original method, chemiluminescent detection, whole blood PCR, two-stage PCR using restriction enzymes Mnl I and Nla III, use of allele-specific probes, with the introduction of a Taq 1 or a Hind III recognition site, in combination with Elisa, or with capillary electrophoresis. A PCR assay using allele-specific primers after microwave irradiation of leukocytes is a novel method which eliminates the need to extract the genomic DNA. The determination of FV:Q506 can also be achieved by using the direct RNA amplification technique (NASBA), together with the detection procedure ELGA (enzyme-linked gel assay).
Summary
The laboratory determination of APC resistance is based on a clotting assay that measures two APTT-reactions, one performed in the presence of exogenous APC and the other in its absence. To obtain reliable results with the classic APC resistance test method, it is necessary for the basal APTT reaction to be within the normal range (25-40 sec). If results from different laboratories are to be compared, it may be beneficial to normalize the APC ratio against the APC ratio of a normal plasma pool. A modification of the classic test is now available, which involves a 1+4 predilution of samples with factor V deficient plasma. This assay variant not only gives a 100% discrimination for the FV:Q506 mutation, but also strongly reduces the influence of preanalytical variations such as plasma handling and storage. Since predilution in factor V-deficient plasma normalizes the basal APTT it safely allows for testing of patients on anticoagulant therapies. DNA-based methods for the detection of the FV:Q506 mutation include primarily PCR amplification of the defect region from genomic material followed by the restriction enzyme cleavage.
